AJHG Volume 99, Issue 1, p163–173, 7 July 2016
Human Y Chromosome Haplogroup N: A Non-trivial Time-Resolved Phylogeography that Cuts across Language Families
Anne-Mai Ilumäe et al.
The paternal haplogroup (hg) N is distributed from southeast Asia to eastern Europe. The demographic processes that have shaped the vast extent of this major Y chromosome lineage across numerous linguistically and autosomally divergent populations have previously been unresolved. On the basis of 94 high-coverage re-sequenced Y chromosomes, we establish and date a detailed hg N phylogeny. We evaluate geographic structure by using 16 distinguishing binary markers in 1,631 hg N Y chromosomes from a collection of 6,521 samples from 56 populations. The more southerly distributed sub-clade N4 emerged before N2a1 and N3, found mostly in the north, but the latter two display more elaborate branching patterns, indicative of regional contrasts in recent expansions. In particular, a number of prominent and well-defined clades with common N3a3’6 ancestry occur in regionally dissimilar northern Eurasian populations, indicating almost simultaneous regional diversification and expansion within the last 5,000 years. This patrilineal genetic affinity is decoupled from the associated higher degree of language diversity.
Link

Showing posts with label Y chromosome. Show all posts
Showing posts with label Y chromosome. Show all posts
July 11, 2016
April 25, 2016
Bursts in human male demography

When the tree is calibrated with a mutation rate estimate of 0.76 × 10-9 mutations per base pair per year9, the time to the most recent common ancestor (TMRCA) of the tree is ~190,000 years, but we consider the implications of alternative mutation rate estimates below. Of the clades resulting from the four deepest branching events, all but one are exclusive to Africa, and the TMRCA of all non-African lineages (that is, the TMRCA of haplogroups DE and CF) is ~76,000 years (Fig. 1, Supplementary Figs. 18 and 19, Supplementary Table 10, and Supplementary Note). We saw a notable increase in the number of lineages outside Africa ~50–55 kya, perhaps reflecting the geographical expansion and differentiation of Eurasian populations as they settled the vast expanse of these continents. Consistent with previous proposals14, a parsimonious interpretation of the phylogeny is that the predominant African haplogroup, haplogroup E, arose outside the continent. This model of geographical segregation within the CT clade requires just one continental haplogroup exchange (E to Africa), rather than three (D, C, and F out of Africa). Furthermore, the timing of this putative return to Africa—between the emergence of haplogroup E and its differentiation within Africa by 58 kya—is consistent with proposals, based on non–Y chromosome data, of abundant gene flow between Africa and nearby regions of Asia 50–80 kya15.I've long argued for the Y-chromosome haplogroup E migration into Africa and it is nice to see this common-sense interpretation finally adopted. Too much focus has been placed on figuring out which routes modern humans took out of Africa, and not at all to figure out how Eurasian males came to overwhelm the African Y-chromosome gene pool so decisively. The Eurasian migration into Africa must have taken place in the ~70-60kya window, constrained by the D/E split and the deepest intra-African E splits. I think that the Out-of-Arabia scenario I outlined in 2012 continues to make a lot of sense. It would be awesome to get data from the first Later Stone Age people from Africa which are probably the best bet to trace this migration from Eurasia into Sub-Saharan Africa.

Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences
G David Poznik et al.
We report the sequences of 1,244 human Y chromosomes randomly ascertained from 26 worldwide populations by the 1000 Genomes Project. We discovered more than 65,000 variants, including single-nucleotide variants, multiple-nucleotide variants, insertions and deletions, short tandem repeats, and copy number variants. Of these, copy number variants contribute the greatest predicted functional impact. We constructed a calibrated phylogenetic tree on the basis of binary single-nucleotide variants and projected the more complex variants onto it, estimating the number of mutations for each class. Our phylogeny shows bursts of extreme expansion in male numbers that have occurred independently among each of the five continental superpopulations examined, at times of known migrations and technological innovations.
Link
April 07, 2016
Neandertal Y-chromosome (finally)
It's been six years since the publication of the draft Neandertal genome, and one piece of the puzzle that's always been missing is the Neandertal Y-chromosome (unfortunately most of the Neandertals yielding data were female). But, the wait is finally over, with the first publication of Neandertal Y-chromosome data.
AJHG Volume 98, Issue 4, p728–734, 7 April 2016
The Divergence of Neandertal and Modern Human Y Chromosomes
Fernando L. Mendez, G. David Poznik, Sergi Castellano, Carlos D. Bustamante
Sequencing the genomes of extinct hominids has reshaped our understanding of modern human origins. Here, we analyze ∼120 kb of exome-captured Y-chromosome DNA from a Neandertal individual from El Sidrón, Spain. We investigate its divergence from orthologous chimpanzee and modern human sequences and find strong support for a model that places the Neandertal lineage as an outgroup to modern human Y chromosomes—including A00, the highly divergent basal haplogroup. We estimate that the time to the most recent common ancestor (TMRCA) of Neandertal and modern human Y chromosomes is ∼588 thousand years ago (kya) (95% confidence interval [CI]: 447–806 kya). This is ∼2.1 (95% CI: 1.7–2.9) times longer than the TMRCA of A00 and other extant modern human Y-chromosome lineages. This estimate suggests that the Y-chromosome divergence mirrors the population divergence of Neandertals and modern human ancestors, and it refutes alternative scenarios of a relatively recent or super-archaic origin of Neandertal Y chromosomes. The fact that the Neandertal Y we describe has never been observed in modern humans suggests that the lineage is most likely extinct. We identify protein-coding differences between Neandertal and modern human Y chromosomes, including potentially damaging changes to PCDH11Y, TMSB4Y, USP9Y, and KDM5D. Three of these changes are missense mutations in genes that produce male-specific minor histocompatibility (H-Y) antigens. Antigens derived from KDM5D, for example, are thought to elicit a maternal immune response during gestation. It is possible that incompatibilities at one or more of these genes played a role in the reproductive isolation of the two groups.
Link
AJHG Volume 98, Issue 4, p728–734, 7 April 2016
The Divergence of Neandertal and Modern Human Y Chromosomes
Fernando L. Mendez, G. David Poznik, Sergi Castellano, Carlos D. Bustamante
Sequencing the genomes of extinct hominids has reshaped our understanding of modern human origins. Here, we analyze ∼120 kb of exome-captured Y-chromosome DNA from a Neandertal individual from El Sidrón, Spain. We investigate its divergence from orthologous chimpanzee and modern human sequences and find strong support for a model that places the Neandertal lineage as an outgroup to modern human Y chromosomes—including A00, the highly divergent basal haplogroup. We estimate that the time to the most recent common ancestor (TMRCA) of Neandertal and modern human Y chromosomes is ∼588 thousand years ago (kya) (95% confidence interval [CI]: 447–806 kya). This is ∼2.1 (95% CI: 1.7–2.9) times longer than the TMRCA of A00 and other extant modern human Y-chromosome lineages. This estimate suggests that the Y-chromosome divergence mirrors the population divergence of Neandertals and modern human ancestors, and it refutes alternative scenarios of a relatively recent or super-archaic origin of Neandertal Y chromosomes. The fact that the Neandertal Y we describe has never been observed in modern humans suggests that the lineage is most likely extinct. We identify protein-coding differences between Neandertal and modern human Y chromosomes, including potentially damaging changes to PCDH11Y, TMSB4Y, USP9Y, and KDM5D. Three of these changes are missense mutations in genes that produce male-specific minor histocompatibility (H-Y) antigens. Antigens derived from KDM5D, for example, are thought to elicit a maternal immune response during gestation. It is possible that incompatibilities at one or more of these genes played a role in the reproductive isolation of the two groups.
Link
February 26, 2016
No Y-chromosomes of recent Indian origin in Australians
Current Biology http://dx.doi.org/10.1016/j.cub.2016.01.028
Deep Roots for Aboriginal Australian Y Chromosomes
Anders Bergström et al.
Australia was one of the earliest regions outside Africa to be colonized by fully modern humans, with archaeological evidence for human presence by 47,000 years ago (47 kya) widely accepted [ 1, 2 ]. However, the extent of subsequent human entry before the European colonial age is less clear. The dingo reached Australia about 4 kya, indirectly implying human contact, which some have linked to changes in language and stone tool technology to suggest substantial cultural changes at the same time [ 3 ]. Genetic data of two kinds have been proposed to support gene flow from the Indian subcontinent to Australia at this time, as well: first, signs of South Asian admixture in Aboriginal Australian genomes have been reported on the basis of genome-wide SNP data [ 4 ]; and second, a Y chromosome lineage designated haplogroup C∗, present in both India and Australia, was estimated to have a most recent common ancestor around 5 kya and to have entered Australia from India [ 5 ]. Here, we sequence 13 Aboriginal Australian Y chromosomes to re-investigate their divergence times from Y chromosomes in other continents, including a comparison of Aboriginal Australian and South Asian haplogroup C chromosomes. We find divergence times dating back to ∼50 kya, thus excluding the Y chromosome as providing evidence for recent gene flow from India into Australia.
Link
Deep Roots for Aboriginal Australian Y Chromosomes
Anders Bergström et al.
Australia was one of the earliest regions outside Africa to be colonized by fully modern humans, with archaeological evidence for human presence by 47,000 years ago (47 kya) widely accepted [ 1, 2 ]. However, the extent of subsequent human entry before the European colonial age is less clear. The dingo reached Australia about 4 kya, indirectly implying human contact, which some have linked to changes in language and stone tool technology to suggest substantial cultural changes at the same time [ 3 ]. Genetic data of two kinds have been proposed to support gene flow from the Indian subcontinent to Australia at this time, as well: first, signs of South Asian admixture in Aboriginal Australian genomes have been reported on the basis of genome-wide SNP data [ 4 ]; and second, a Y chromosome lineage designated haplogroup C∗, present in both India and Australia, was estimated to have a most recent common ancestor around 5 kya and to have entered Australia from India [ 5 ]. Here, we sequence 13 Aboriginal Australian Y chromosomes to re-investigate their divergence times from Y chromosomes in other continents, including a comparison of Aboriginal Australian and South Asian haplogroup C chromosomes. We find divergence times dating back to ∼50 kya, thus excluding the Y chromosome as providing evidence for recent gene flow from India into Australia.
Link
December 29, 2015
Bronze Age people from Ireland had steppe ancestry and R1b

We were able to deduce that Neolithic Ballynahatty had a dark hair shade (99.5% probability), most likely black (86.1% probability), and brown eyes (97.3% probability) (46). Bronze Age Rathlin1 probably had a light hair shade (61.4%) and brown eyes (64.3%). However, each Rathlin genome possessed indication of at least one copy of a haplotype associated with blue eye color in the HERC2/OCA2 region.and:
Third, we followed the methods described in Haak et al. (9), which use a collection of outgroup populations, to estimate the mixture proportions of three different sources, Linearbandkeramik (Early Neolithic; 35 ± 6%), Loschbour (WHG; 26 ± 12%), and Yamnaya (39 ± 8%), in the total Irish Bronze Age group. These three approaches give an overlapping estimate of ∼32% Yamnaya ancestry.PNAS doi: 10.1073/pnas.1518445113
Neolithic and Bronze Age migration to Ireland and establishment of the insular Atlantic genome
Lara M. Cassidy, Rui Martiniano et al.
The Neolithic and Bronze Age transitions were profound cultural shifts catalyzed in parts of Europe by migrations, first of early farmers from the Near East and then Bronze Age herders from the Pontic Steppe. However, a decades-long, unresolved controversy is whether population change or cultural adoption occurred at the Atlantic edge, within the British Isles. We address this issue by using the first whole genome data from prehistoric Irish individuals. A Neolithic woman (3343–3020 cal BC) from a megalithic burial (10.3× coverage) possessed a genome of predominantly Near Eastern origin. She had some hunter–gatherer ancestry but belonged to a population of large effective size, suggesting a substantial influx of early farmers to the island. Three Bronze Age individuals from Rathlin Island (2026–1534 cal BC), including one high coverage (10.5×) genome, showed substantial Steppe genetic heritage indicating that the European population upheavals of the third millennium manifested all of the way from southern Siberia to the western ocean. This turnover invites the possibility of accompanying introduction of Indo-European, perhaps early Celtic, language. Irish Bronze Age haplotypic similarity is strongest within modern Irish, Scottish, and Welsh populations, and several important genetic variants that today show maximal or very high frequencies in Ireland appear at this horizon. These include those coding for lactase persistence, blue eye color, Y chromosome R1b haplotypes, and the hemochromatosis C282Y allele; to our knowledge, the first detection of a known Mendelian disease variant in prehistory. These findings together suggest the establishment of central attributes of the Irish genome 4,000 y ago.
Link
December 22, 2015
Refining Y-chromosome phylogeny with South African sequences
bioRxiv http://dx.doi.org/10.1101/034983
Refining the Y chromosome phylogeny with southern African sequences
Chiara Barbieri, Alexander Hübner, Enrico Macholdt, Shengyu Ni, Sebastian Lippold, Roland Schröder, Sununguko Wata Mpoloka, Josephine Purps, Lutz Roewer, Mark Stoneking, Brigitte Pakendorf
The recent availability of large-scale sequence data for the human Y chromosome has revolutionized analyses of and insights gained from this non-recombining, paternally inherited chromosome. However, the studies to date focus on Eurasian variation, and hence the diversity of early-diverging branches found in Africa has not been adequately documented. Here we analyze over 900 kb of Y chromosome sequence obtained from 547 individuals from southern African Khoisan and Bantu-speaking populations, identifying 232 new sequences from basal haplogroups A and B. We find new branches within haplogroups A2 and A3b1 and suggest that the prehistory of haplogroup B2a is more complex than previously suspected; this haplogroup is likely to have existed in Khoisan groups before the arrival of Bantu-speakers, who brought additional B2a lineages to southern Africa. Furthermore, we estimate older dates than obtained previously for both the A2-T node within the human Y chromosome phylogeny and for some individual haplogroups. Finally, there is pronounced variation in branch length between major haplogroups; haplogroups associated with Bantu-speakers have significantly longer branches. This likely reflects a combination of biases in the SNP calling process and demographic factors, such as an older average paternal age (hence a higher mutation rate), a higher effective population size, and/or a stronger effect of population expansion for Bantu-speakers than for Khoisan groups.
Link
Refining the Y chromosome phylogeny with southern African sequences
Chiara Barbieri, Alexander Hübner, Enrico Macholdt, Shengyu Ni, Sebastian Lippold, Roland Schröder, Sununguko Wata Mpoloka, Josephine Purps, Lutz Roewer, Mark Stoneking, Brigitte Pakendorf
The recent availability of large-scale sequence data for the human Y chromosome has revolutionized analyses of and insights gained from this non-recombining, paternally inherited chromosome. However, the studies to date focus on Eurasian variation, and hence the diversity of early-diverging branches found in Africa has not been adequately documented. Here we analyze over 900 kb of Y chromosome sequence obtained from 547 individuals from southern African Khoisan and Bantu-speaking populations, identifying 232 new sequences from basal haplogroups A and B. We find new branches within haplogroups A2 and A3b1 and suggest that the prehistory of haplogroup B2a is more complex than previously suspected; this haplogroup is likely to have existed in Khoisan groups before the arrival of Bantu-speakers, who brought additional B2a lineages to southern Africa. Furthermore, we estimate older dates than obtained previously for both the A2-T node within the human Y chromosome phylogeny and for some individual haplogroups. Finally, there is pronounced variation in branch length between major haplogroups; haplogroups associated with Bantu-speakers have significantly longer branches. This likely reflects a combination of biases in the SNP calling process and demographic factors, such as an older average paternal age (hence a higher mutation rate), a higher effective population size, and/or a stronger effect of population expansion for Bantu-speakers than for Khoisan groups.
Link
July 12, 2015
Phylogeographic refinement of haplogroup E
Genome Biol Evol (2015) 7 (7): 1940-1950.
Phylogeographic Refinement and Large Scale Genotyping of Human Y Chromosome Haplogroup E Provide New Insights into the Dispersal of Early Pastoralists in the African Continent
Beniamino Trombetta et al.
Haplogroup E, defined by mutation M40, is the most common human Y chromosome clade within Africa. To increase the level of resolution of haplogroup E, we disclosed the phylogenetic relationships among 729 mutations found in 33 haplogroup DE Y-chromosomes sequenced at high coverage in previous studies. Additionally, we dissected the E-M35 subclade by genotyping 62 informative markers in 5,222 samples from 118 worldwide populations. The phylogeny of haplogroup E showed novel features compared with the previous topology, including a new basal dichotomy. Within haplogroup E-M35, we resolved all the previously known polytomies and assigned all the E-M35* chromosomes to five new different clades, all belonging to a newly identified subhaplogroup (E-V1515), which accounts for almost half of the E-M35 chromosomes from the Horn of Africa. Moreover, using a Bayesian phylogeographic analysis and a single nucleotide polymorphism-based approach we localized and dated the origin of this new lineage in the northern part of the Horn, about 12 ka. Time frames, phylogenetic structuring, and sociogeographic distribution of E-V1515 and its subclades are consistent with a multistep demic spread of pastoralism within north-eastern Africa and its subsequent diffusion to subequatorial areas. In addition, our results increase the discriminative power of the E-M35 haplogroup for use in forensic genetics through the identification of new ancestry-informative markers.
Link
Phylogeographic Refinement and Large Scale Genotyping of Human Y Chromosome Haplogroup E Provide New Insights into the Dispersal of Early Pastoralists in the African Continent
Beniamino Trombetta et al.
Haplogroup E, defined by mutation M40, is the most common human Y chromosome clade within Africa. To increase the level of resolution of haplogroup E, we disclosed the phylogenetic relationships among 729 mutations found in 33 haplogroup DE Y-chromosomes sequenced at high coverage in previous studies. Additionally, we dissected the E-M35 subclade by genotyping 62 informative markers in 5,222 samples from 118 worldwide populations. The phylogeny of haplogroup E showed novel features compared with the previous topology, including a new basal dichotomy. Within haplogroup E-M35, we resolved all the previously known polytomies and assigned all the E-M35* chromosomes to five new different clades, all belonging to a newly identified subhaplogroup (E-V1515), which accounts for almost half of the E-M35 chromosomes from the Horn of Africa. Moreover, using a Bayesian phylogeographic analysis and a single nucleotide polymorphism-based approach we localized and dated the origin of this new lineage in the northern part of the Horn, about 12 ka. Time frames, phylogenetic structuring, and sociogeographic distribution of E-V1515 and its subclades are consistent with a multistep demic spread of pastoralism within north-eastern Africa and its subsequent diffusion to subequatorial areas. In addition, our results increase the discriminative power of the E-M35 haplogroup for use in forensic genetics through the identification of new ancestry-informative markers.
Link
Y-chromosomes of Sicilian and Calabrian Arbereshe
European Journal of Human Genetics advance online publication 1 July 2015; doi: 10.1038/ejhg.2015.138
Shared language, diverging genetic histories: high-resolution analysis of Y-chromosome variability in Calabrian and Sicilian Arbereshe
Stefania Sarno et al.
The relationship between genetic and linguistic diversification in human populations has been often explored to interpret some specific issues in human history. The Albanian-speaking minorities of Sicily and Southern Italy (Arbereshe) constitute an important portion of the ethnolinguistic variability of Italy. Their linguistic isolation from neighboring Italian populations and their documented migration history, make such minorities particularly effective for investigating the interplay between cultural, geographic and historical factors. Nevertheless, the extent of Arbereshe genetic relationships with the Balkan homeland and the Italian recipient populations has been only partially investigated. In the present study we address the genetic history of Arbereshe people by combining highly resolved analyses of Y-chromosome lineages and extensive computer simulations. A large set of slow- and fast-evolving molecular markers was typed in different Arbereshe communities from Sicily and Southern Italy (Calabria), as well as in both the putative Balkan source and Italian sink populations. Our results revealed that the considered Arbereshe groups, despite speaking closely related languages and sharing common cultural features, actually experienced diverging genetic histories. The estimated proportions of genetic admixture confirm the tight relationship of Calabrian Arbereshe with modern Albanian populations, in accordance with linguistic hypotheses. On the other hand, population stratification and/or an increased permeability of linguistic and geographic barriers may be hypothesized for Sicilian groups, to account for their partial similarity with Greek populations and their higher levels of local admixture. These processes ultimately resulted in the differential acquisition or preservation of specific paternal lineages by the present-day Arbereshe communities.
Link
Shared language, diverging genetic histories: high-resolution analysis of Y-chromosome variability in Calabrian and Sicilian Arbereshe
Stefania Sarno et al.
The relationship between genetic and linguistic diversification in human populations has been often explored to interpret some specific issues in human history. The Albanian-speaking minorities of Sicily and Southern Italy (Arbereshe) constitute an important portion of the ethnolinguistic variability of Italy. Their linguistic isolation from neighboring Italian populations and their documented migration history, make such minorities particularly effective for investigating the interplay between cultural, geographic and historical factors. Nevertheless, the extent of Arbereshe genetic relationships with the Balkan homeland and the Italian recipient populations has been only partially investigated. In the present study we address the genetic history of Arbereshe people by combining highly resolved analyses of Y-chromosome lineages and extensive computer simulations. A large set of slow- and fast-evolving molecular markers was typed in different Arbereshe communities from Sicily and Southern Italy (Calabria), as well as in both the putative Balkan source and Italian sink populations. Our results revealed that the considered Arbereshe groups, despite speaking closely related languages and sharing common cultural features, actually experienced diverging genetic histories. The estimated proportions of genetic admixture confirm the tight relationship of Calabrian Arbereshe with modern Albanian populations, in accordance with linguistic hypotheses. On the other hand, population stratification and/or an increased permeability of linguistic and geographic barriers may be hypothesized for Sicilian groups, to account for their partial similarity with Greek populations and their higher levels of local admixture. These processes ultimately resulted in the differential acquisition or preservation of specific paternal lineages by the present-day Arbereshe communities.
Link
May 21, 2015
More Y-chromosome super-fathers

 There may be additional founders with recent time depths than shown in the table, e.g., a very shallow clusters within E-M35 (probably E-V13?) and a couple of shallow clusters within I-P215
There may be additional founders with recent time depths than shown in the table, e.g., a very shallow clusters within E-M35 (probably E-V13?) and a couple of shallow clusters within I-P215
The plots are consistent with patterns seen in the relative numbers of singletons, described above, in that the Saami and Palestinians show markedly different demographic histories compared with the rest, featuring very recent reductions, while the Turks and Greeks show evidence of general expansion, with increased growth rate around 14 KYA. A different pattern is seen in the remaining majority (13/17) of populations, which share remarkably similar histories featuring a minimum effective population size ~2.1–4.2 KYA (considering the 95% confidence intervals (CIs) reported in Supplementary Table 4), followed by expansion to the present.
Related:
Nature Communications 6, Article number: 7152 doi:10.1038/ncomms8152
Large-scale recent expansion of European patrilineages shown by population resequencing
Chiara Batini, Pille Hallast et al.
The proportion of Europeans descending from Neolithic farmers ~10 thousand years ago (KYA) or Palaeolithic hunter-gatherers has been much debated. The male-specific region of the Y chromosome (MSY) has been widely applied to this question, but unbiased estimates of diversity and time depth have been lacking. Here we show that European patrilineages underwent a recent continent-wide expansion. Resequencing of 3.7 Mb of MSY DNA in 334 males, comprising 17 European and Middle Eastern populations, defines a phylogeny containing 5,996 single-nucleotide polymorphisms. Dating indicates that three major lineages (I1, R1a and R1b), accounting for 64% of our sample, have very recent coalescent times, ranging between 3.5 and 7.3 KYA. A continuous swathe of 13/17 populations share similar histories featuring a demographic expansion starting ~2.1–4.2 KYA. Our results are compatible with ancient MSY DNA data, and contrast with data on mitochondrial DNA, indicating a widespread male-specific phenomenon that focuses interest on the social structure of Bronze Age Europe.
Link
May 03, 2015
Structure of Y-haplogroup N
arXiv:1504.06463 [q-bio.PE]
The dichotomy structure of Y chromosome Haplogroup N
Kang Hu et al.
Haplogroup N-M231 of human Y chromosome is a common clade from Eastern Asia to Northern Europe, being one of the most frequent haplogroups in Altaic and Uralic-speaking populations. Using newly discovered bi-allelic markers from high-throughput DNA sequencing, we largely improved the phylogeny of Haplogroup N, in which 16 subclades could be identified by 33 SNPs. More than 400 males belonging to Haplogroup N in 34 populations in China were successfully genotyped, and populations in Northern Asia and Eastern Europe were also compared together. We found that all the N samples were typed as inside either clade N1-F1206 (including former N1a-M128, N1b-P43 and N1c-M46 clades), most of which were found in Altaic, Uralic, Russian and Chinese-speaking populations, or N2-F2930, common in Tibeto-Burman and Chinese-speaking populations. Our detailed results suggest that Haplogroup N developed in the region of China since the final stage of late Paleolithic Era.
Link
The dichotomy structure of Y chromosome Haplogroup N
Kang Hu et al.
Haplogroup N-M231 of human Y chromosome is a common clade from Eastern Asia to Northern Europe, being one of the most frequent haplogroups in Altaic and Uralic-speaking populations. Using newly discovered bi-allelic markers from high-throughput DNA sequencing, we largely improved the phylogeny of Haplogroup N, in which 16 subclades could be identified by 33 SNPs. More than 400 males belonging to Haplogroup N in 34 populations in China were successfully genotyped, and populations in Northern Asia and Eastern Europe were also compared together. We found that all the N samples were typed as inside either clade N1-F1206 (including former N1a-M128, N1b-P43 and N1c-M46 clades), most of which were found in Altaic, Uralic, Russian and Chinese-speaking populations, or N2-F2930, common in Tibeto-Burman and Chinese-speaking populations. Our detailed results suggest that Haplogroup N developed in the region of China since the final stage of late Paleolithic Era.
Link
April 13, 2015
Haplogroup G1, Y-chromosome mutation rate and migrations of Iranic speakers

I am guessing that the story of Iranian origins will only be solved in correlation to their Indo-Aryan brethren and their more distant Indo-European relations.
Clearly, G1 cannot be Proto-Indo-European as it has a rather limited distribution in Eurasia, but it could very well have been a marker of a subset of Indo-Europeans. If it was present in ancestral Iranians, then this would geographically constrain the places where ancestral Iranians were formed.
PLoS ONE 10(4): e0122968. doi:10.1371/journal.pone.0122968
Deep Phylogenetic Analysis of Haplogroup G1 Provides Estimates of SNP and STR Mutation Rates on the Human Y-Chromosome and Reveals Migrations of Iranic Speakers
Oleg Balanovsky et al.
Y-chromosomal haplogroup G1 is a minor component of the overall gene pool of South-West and Central Asia but reaches up to 80% frequency in some populations scattered within this area. We have genotyped the G1-defining marker M285 in 27 Eurasian populations (n= 5,346), analyzed 367 M285-positive samples using 17 Y-STRs, and sequenced ~11 Mb of the Y-chromosome in 20 of these samples to an average coverage of 67X. This allowed detailed phylogenetic reconstruction. We identified five branches, all with high geographical specificity: G1-L1323 in Kazakhs, the closely related G1-GG1 in Mongols, G1-GG265 in Armenians and its distant brother clade G1-GG162 in Bashkirs, and G1-GG362 in West Indians. The haplotype diversity, which decreased from West Iran to Central Asia, allows us to hypothesize that this rare haplogroup could have been carried by the expansion of Iranic speakers northwards to the Eurasian steppe and via founder effects became a predominant genetic component of some populations, including the Argyn tribe of the Kazakhs. The remarkable agreement between genetic and genealogical trees of Argyns allowed us to calibrate the molecular clock using a historical date (1405 AD) of the most recent common genealogical ancestor. The mutation rate for Y-chromosomal sequence data obtained was 0.78×10-9 per bp per year, falling within the range of published rates. The mutation rate for Y-chromosomal STRs was 0.0022 per locus per generation, very close to the so-called genealogical rate. The “clan-based” approach to estimating the mutation rate provides a third, middle way between direct farther-to-son comparisons and using archeologically known migrations, whose dates are subject to revision and of uncertain relationship to genetic events.
Link
March 25, 2015
Icelanders galore
A set of four papers in Nature Genetics today. All open access. Of interest from the Y-chromosome paper:
Nature Genetics (2015) doi:10.1038/ng.3247
Large-scale whole-genome sequencing of the Icelandic population
Daniel F Gudbjartsson et al.
Here we describe the insights gained from sequencing the whole genomes of 2,636 Icelanders to a median depth of 20×. We found 20 million SNPs and 1.5 million insertions-deletions (indels). We describe the density and frequency spectra of sequence variants in relation to their functional annotation, gene position, pathway and conservation score. We demonstrate an excess of homozygosity and rare protein-coding variants in Iceland. We imputed these variants into 104,220 individuals down to a minor allele frequency of 0.1% and found a recessive frameshift mutation in MYL4 that causes early-onset atrial fibrillation, several mutations in ABCB4 that increase risk of liver diseases and an intronic variant in GNAS associating with increased thyroid-stimulating hormone levels when maternally inherited. These data provide a study design that can be used to determine how variation in the sequence of the human genome gives rise to human diversity.
Link
Nature Genetics (2015) doi:10.1038/ng.3171
The Y-chromosome point mutation rate in humans
Agnar Helgason et al.
Mutations are the fundamental source of biological variation, and their rate is a crucial parameter for evolutionary and medical studies. Here we used whole-genome sequence data from 753 Icelandic males, grouped into 274 patrilines, to estimate the point mutation rate for 21.3 Mb of male-specific Y chromosome (MSY) sequence, on the basis of 1,365 meioses (47,123 years). The combined mutation rate for 15.2 Mb of X-degenerate (XDG), X-transposed (XTR) and ampliconic excluding palindromes (rAMP) sequence was 8.71 × 10−10 mutations per position per year (PPPY). We observed a lower rate (P = 0.04) of 7.37 × 10−10 PPPY for 6.1 Mb of sequence from palindromes (PAL), which was not statistically different from the rate of 7.2 × 10−10 PPPY for paternally transmitted autosomes1. We postulate that the difference between PAL and the other MSY regions may provide an indication of the rate at which nascent autosomal and PAL de novo mutations are repaired as a result of gene conversion.
Link
Nature Genetics (2015) doi:10.1038/ng.3246
Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease
Stacy Steinberg et al.
We conducted a search for rare, functional variants altering susceptibility to Alzheimer's disease that exploited knowledge of common variants associated with the same disease. We found that loss-of-function variants in ABCA7 confer risk of Alzheimer's disease in Icelanders (odds ratio (OR) = 2.12, P = 2.2 × 10−13) and discovered that the association replicated in study groups from Europe and the United States (combined OR = 2.03, P = 6.8 × 10−15).
Link
Nature Genetics (2015) doi:10.1038/ng.3243
Identification of a large set of rare complete human knockouts
Patrick Sulem et al.
Loss-of-function mutations cause many mendelian diseases. Here we aimed to create a catalog of autosomal genes that are completely knocked out in humans by rare loss-of-function mutations. We sequenced the whole genomes of 2,636 Icelanders and imputed the sequence variants identified in this set into 101,584 additional chip-genotyped and phased Icelanders. We found a total of 6,795 autosomal loss-of-function SNPs and indels in 4,924 genes. Of the genotyped Icelanders, 7.7% are homozygotes or compound heterozygotes for loss-of-function mutations with a minor allele frequency (MAF) below 2% in 1,171 genes (complete knockouts). Genes that are highly expressed in the brain are less often completely knocked out than other genes. Homozygous loss-of-function offspring of two heterozygous parents occurred less frequently than expected (deficit of 136 per 10,000 transmissions for variants with MAF less than 2%, 95% confidence interval (CI) = 10–261).
Link
When this rate was applied to estimate the TMRCA between two Y chromosomes that encompass the oldest known patrilineal bifurcation between any humans (representing haplogroups A00 and A0, with 75 derived mutational differences in 180 kb of XDG sequence)19, we obtained a maximum-likelihood estimate21 of 239,000 years ago and a 95% CI of 188,000–296,000 years ago (174,000–321,000 years ago when incorporating the 95% CI of our mutation rate).This seems similar to the 254kya estimated by Karmin et al.
Nature Genetics (2015) doi:10.1038/ng.3247
Large-scale whole-genome sequencing of the Icelandic population
Daniel F Gudbjartsson et al.
Here we describe the insights gained from sequencing the whole genomes of 2,636 Icelanders to a median depth of 20×. We found 20 million SNPs and 1.5 million insertions-deletions (indels). We describe the density and frequency spectra of sequence variants in relation to their functional annotation, gene position, pathway and conservation score. We demonstrate an excess of homozygosity and rare protein-coding variants in Iceland. We imputed these variants into 104,220 individuals down to a minor allele frequency of 0.1% and found a recessive frameshift mutation in MYL4 that causes early-onset atrial fibrillation, several mutations in ABCB4 that increase risk of liver diseases and an intronic variant in GNAS associating with increased thyroid-stimulating hormone levels when maternally inherited. These data provide a study design that can be used to determine how variation in the sequence of the human genome gives rise to human diversity.
Link
Nature Genetics (2015) doi:10.1038/ng.3171
The Y-chromosome point mutation rate in humans
Agnar Helgason et al.
Mutations are the fundamental source of biological variation, and their rate is a crucial parameter for evolutionary and medical studies. Here we used whole-genome sequence data from 753 Icelandic males, grouped into 274 patrilines, to estimate the point mutation rate for 21.3 Mb of male-specific Y chromosome (MSY) sequence, on the basis of 1,365 meioses (47,123 years). The combined mutation rate for 15.2 Mb of X-degenerate (XDG), X-transposed (XTR) and ampliconic excluding palindromes (rAMP) sequence was 8.71 × 10−10 mutations per position per year (PPPY). We observed a lower rate (P = 0.04) of 7.37 × 10−10 PPPY for 6.1 Mb of sequence from palindromes (PAL), which was not statistically different from the rate of 7.2 × 10−10 PPPY for paternally transmitted autosomes1. We postulate that the difference between PAL and the other MSY regions may provide an indication of the rate at which nascent autosomal and PAL de novo mutations are repaired as a result of gene conversion.
Link
Nature Genetics (2015) doi:10.1038/ng.3246
Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease
Stacy Steinberg et al.
We conducted a search for rare, functional variants altering susceptibility to Alzheimer's disease that exploited knowledge of common variants associated with the same disease. We found that loss-of-function variants in ABCA7 confer risk of Alzheimer's disease in Icelanders (odds ratio (OR) = 2.12, P = 2.2 × 10−13) and discovered that the association replicated in study groups from Europe and the United States (combined OR = 2.03, P = 6.8 × 10−15).
Link
Nature Genetics (2015) doi:10.1038/ng.3243
Identification of a large set of rare complete human knockouts
Patrick Sulem et al.
Loss-of-function mutations cause many mendelian diseases. Here we aimed to create a catalog of autosomal genes that are completely knocked out in humans by rare loss-of-function mutations. We sequenced the whole genomes of 2,636 Icelanders and imputed the sequence variants identified in this set into 101,584 additional chip-genotyped and phased Icelanders. We found a total of 6,795 autosomal loss-of-function SNPs and indels in 4,924 genes. Of the genotyped Icelanders, 7.7% are homozygotes or compound heterozygotes for loss-of-function mutations with a minor allele frequency (MAF) below 2% in 1,171 genes (complete knockouts). Genes that are highly expressed in the brain are less often completely knocked out than other genes. Homozygous loss-of-function offspring of two heterozygous parents occurred less frequently than expected (deficit of 136 per 10,000 transmissions for variants with MAF less than 2%, 95% confidence interval (CI) = 10–261).
Link
March 14, 2015
Bottleneck in human Y-chromosomes in the last 10,000 years.
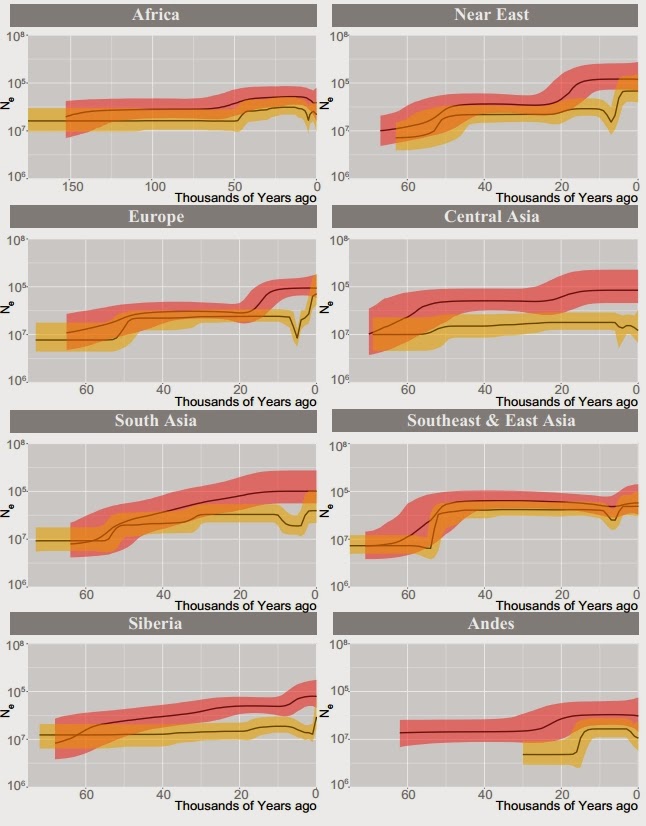
The contrast (left) between mtDNA (red) and Y-chromosome (yellow) coalescences is quite noticeable. The little "dip" in the yellow curve in many regions on the right of the various regional plots corresponds to a the second bottleneck event (that was really not "one" event, but rather shows that many modern men descend from a small number of "patriarchs" of the Neolithic and Bronze Age worlds. The "when" of the dip is important:

Most human mythologies contain stories of "first men" and eponymous founders of nations; these were often ridiculed in recent times as invented stories whose purpose was to engender social cohesion through a story of shared descent. But, now it seems that these stories were at least in part true, and such ultra-prolific patriarchs do indeed stand at the beginning of many later lines of descent.

The split between DT and B2'5 is placed at around ~100 thousand years ago. This corresponds perfectly (in my opinion) to the Out-of-Africa event from which most Eurasian men are probably descended. For the next thirty thousand years, Eurasians were probably confined to Arabia and the Middle East. The next major event is the foundation of the unambiguously Eurasian CT lineage ~70 thousand years ago (coinciding with the Toba eruption and the onset of super arid conditions at the onset of MIS 4). And, the final event of the "grand picture" of Eurasian prehistory was the Upper Paleolithic at ~50 thousand years ago, when Eurasians finally got the "tech" to exhibit complex behavior, invent new tools, conquer diverse environments and ultimately colonize the entire planet while driving Eurasian archaics to extinction.
An important detail in this grand picture is the fact that the authors estimate the coalescence date between D and E1'4 to ~70,000 years, coinciding with the C/GT split from the same time. These lineages are all found in Eurasia, but only E1'4 is found in Africa. I think this points clearly to back-migration of Eurasians into Africa, perhaps as environmental refugees following the c. 70kya Arabian ecological catastrophe. In any case, the fact that two separate Eurasian-specific lineages (CT and D) coalesce to ~70kya destroys the theory that the spread of modern humans into Eurasia happened together with UP-related technologies, a theory that was already on its last legs given the evidence that pre-UP admixture with Neandertals had taken place (as such admixture would have been impossible if pre-UP Eurasians were not already present outside of Africa at that time).
Genome Research doi:10.1101/gr.186684.114
A recent bottleneck of Y chromosome diversity coincides with a global change in culture
Monika Karmin et al.
It is commonly thought that human genetic diversity in non-African populations was shaped primarily by an out-of-Africa dispersal 50–100 thousand yr ago (kya). Here, we present a study of 456 geographically diverse high-coverage Y chromosome sequences, including 299 newly reported samples. Applying ancient DNA calibration, we date the Y-chromosomal most recent common ancestor (MRCA) in Africa at 254 (95% CI 192–307) kya and detect a cluster of major non-African founder haplogroups in a narrow time interval at 47–52 kya, consistent with a rapid initial colonization model of Eurasia and Oceania after the out-of-Africa bottleneck. In contrast to demographic reconstructions based on mtDNA, we infer a second strong bottleneck in Y-chromosome lineages dating to the last 10 ky. We hypothesize that this bottleneck is caused by cultural changes affecting variance of reproductive success among males.
Link
February 26, 2015
Estonian biocentre high coverage Y chromosome sequences and Turkic data
Courtesy of the good people of the Estonian biocentre:
- 307 high coverage human Y chrmosome sequences. Paper to be published soon.
- The Genetic Legacy of the Expansion of Turkic-Speaking Nomads Across Eurasia. Paper in press.
The Y chromosome data seems particularly exciting (there is a spreadsheet of populations in the download directory). One of the weaknesses of the 1000 Genomes data was that it didn't have any populations between Tuscany and East/South Asia, and the new dataset seems to rectify that.
The Turkic dataset is probably the one used for the preprint The Genetic Legacy of the Expansion of Turkic-Speaking Nomads Across Eurasia. Since I overlooked this when it came out last summer, I'll post about it when the paper is published in a journal.
The Turkic dataset is probably the one used for the preprint The Genetic Legacy of the Expansion of Turkic-Speaking Nomads Across Eurasia. Since I overlooked this when it came out last summer, I'll post about it when the paper is published in a journal.
February 22, 2015
Y chromosomes and Catalan surnames
Some really rich data in the supplements.
European Journal of Human Genetics advance online publication 18 February 2015; doi: 10.1038/ejhg.2015.14
Y-chromosome diversity in Catalan surname samples: insights into surname origin and frequency
Neus Solé-Morata et al.
The biological behavior of the Y chromosome, which is paternally inherited, implies that males sharing the same surname may also share a similar Y chromosome. However, socio-cultural factors, such as polyphyletism, non-paternity, adoption, or matrilineal surname transmission, may prevent the joint transmission of the surname and the Y chromosome. By genotyping 17 Y-STRs and 68 SNPs in ~2500 male samples that each carried one of the 50 selected Catalan surnames, we could determine sets of descendants of a common ancestor, the population of origin of the common ancestor, and the date when such a common ancestor lived. Haplotype diversity was positively correlated with surname frequency, that is, rarer surnames showed the strongest signals of coancestry. Introgression rates of Y chromosomes into a surname by non-paternity, adoption, and transmission of the maternal surname were estimated at 1.5−2.6% per generation, with some local variation. Average ages for the founders of the surnames were estimated at ~500 years, suggesting a delay between the origin of surnames (twelfth and thirteenth centuries) and the systematization of their paternal transmission. We have found that, in general, a foreign etymology for a surname does not often result in a non-indigenous origin of surname founders; however, bearers of some surnames with an Arabic etymology show an excess of North African haplotypes. Finally, we estimate that surname prediction from a Y-chromosome haplotype, which may have interesting forensic applications, has a ~60% sensitivity but a 17% false discovery rate.
Link
European Journal of Human Genetics advance online publication 18 February 2015; doi: 10.1038/ejhg.2015.14
Y-chromosome diversity in Catalan surname samples: insights into surname origin and frequency
Neus Solé-Morata et al.
The biological behavior of the Y chromosome, which is paternally inherited, implies that males sharing the same surname may also share a similar Y chromosome. However, socio-cultural factors, such as polyphyletism, non-paternity, adoption, or matrilineal surname transmission, may prevent the joint transmission of the surname and the Y chromosome. By genotyping 17 Y-STRs and 68 SNPs in ~2500 male samples that each carried one of the 50 selected Catalan surnames, we could determine sets of descendants of a common ancestor, the population of origin of the common ancestor, and the date when such a common ancestor lived. Haplotype diversity was positively correlated with surname frequency, that is, rarer surnames showed the strongest signals of coancestry. Introgression rates of Y chromosomes into a surname by non-paternity, adoption, and transmission of the maternal surname were estimated at 1.5−2.6% per generation, with some local variation. Average ages for the founders of the surnames were estimated at ~500 years, suggesting a delay between the origin of surnames (twelfth and thirteenth centuries) and the systematization of their paternal transmission. We have found that, in general, a foreign etymology for a surname does not often result in a non-indigenous origin of surname founders; however, bearers of some surnames with an Arabic etymology show an excess of North African haplotypes. Finally, we estimate that surname prediction from a Y-chromosome haplotype, which may have interesting forensic applications, has a ~60% sensitivity but a 17% false discovery rate.
Link
February 12, 2015
A story of 69 ancient Europeans

Two Near Eastern migrations into Europe
In 2011, I observed that West Eurasian populations were too close (measured by Fst) to allow for long periods of differentiation between them. By implication, there must have been a "common source" of ancestry uniting them, which I placed in a "womb of nations" of the Neolithic Near East. I proposed that migrations out of this core area homogenized West Eurasians, writing:
In Arabia, the migrants would have met aboriginal Arabians, similar to their next door-neighbors in East Africa, undergoing a subtle African shift (Southwest_Asians). In North Africa, they would have encountered denser populations during the favorable conditions of MIS 1, and by absorbing them they would became the Berbers (Northwest_Africans). Their migrations to the southeast brought them into the realm of Indian-leaning people, in the rich agricultural fields of the Mehrgarh and the now deserted oases of Bactria and Margiana. Across the Mediterranean and along the Atlantic facade of Europe, they would have encountered the Mesolithic populations of Europe, and through their blending became the early Neolithic inhabitants of the Mediterranean and Atlantic coasts of Europe (Mediterraneans). And, to the north, from either the Balkans, the Caucasus, or the trans-Caspian region, they would have met the last remaining Proto-Europeoid hunters of the continental zone, becoming the Northern Europeoids who once stretched all the way to the interior of Asia.
The first migration (early Neolithic) is already uncontroversial, but the paper includes data from Spanish early farmers that are also Sardinian- and LBK-like. The "Sardinian" Iceman was no fluke. It is now proven that not only the LBK but also the Spanish Neolithic came from the same expansion of Mediterranean populations which survives in Sardinia. The authors write:
Principal components analysis (PCA) of all ancient individuals along with 777 present-day West Eurasians4 (Fig. 2a, SI5) replicates the positioning of present-day Europeans between the Near East and European hunter-gatherers4,20, and the clustering of early farmers from across Europe with present day Sardinians3,4,27, suggesting that farming expansions across the Mediterranean to Spain and via the Danubian route to Hungary and Germany descended from a common stock.The second migration went into eastern Europe:
The Yamnaya differ from the EHG by sharing fewer alleles with MA1 (|Z|=6.7) suggesting a dilution of ANE ancestry between 5,000-3,000 BCE on the European steppe. This was likely due to admixture of EHG with a population related to present-day Near Easterners, as the most negative f3-statistic in the Yamnaya (giving unambiguous evidence of admixture) is observed when we model them as a mixture of EHG and present-day Near Eastern populations like Armenians (Z = -6.3; SI7).

The Yamna population generally belongs to the European race. It was tall (175.5cm), dolichocephalic, with broad faces of medium height. Among them there were, however, more robust elements with high and wide faces of the proto-Europoid type, and also more gracile individuals with narrow and high faces, probably reflecting contacts with the East Mediterranean type (Kurts 1984: 90).

It seems that the legacy of the early farmers suffered two hits, which is why only in Sardinia and (to a lesser degree) in southern Europe that they have persisted as the major component of ancestry. The first blow came during the Neolithic:
Middle Neolithic Europeans from Germany, Spain, Hungary, and Sweden from the period ~4,000-3,000 BCE are intermediate between the earlier farmers and the WHG, suggesting an increase of WHG ancestry throughout much of Europe.And the coup de grâce after the 5kya mark:
We estimate that these two elements each contributed about half the ancestry each of the Yamnaya (SI6, SI9), explaining why the population turnover inferred using Yamnaya as a source is about twice as high compared to the undiluted EHG. The estimate of Yamnaya related ancestry in the Corded Ware is consistent when using either present populations or ancient Europeans as outgroups (SI9, SI10), and is 73.1 ± 2.2% when both sets are combined (SI10). [...] The magnitude of the population turnover that occurred becomes even more evident if one considers the fact that the steppe migrants may well have mixed with eastern European agriculturalists on their way to central Europe. Thus, we cannot exclude a scenario in which the Corded Ware arriving in today’s Germany had no ancestry at all from local populations.

In 2012 I had used the paltry data on a handful ancient DNA samples to observe that in ADMIXTURE modern Europeans had a West Asian genetic component (peaking in "Caucasus" and "Gedrosia") that pre-5kya Europeans didn't. I proposed that the Bronze Age migration of the Indo-Europeans spread this component:
But there is another component present in modern Europe, the West_Asian which is conspicuous in its absence in all the ancient samples so far. This component reaches its highest occurrence in the highlands of West Asia, from Anatolia and the Caucasus all the way to the Indian subcontinent. [...] Nonetheless, some of the legacy of the earliest Indo-European speakers does appear to persist down to the present day in the genomes of their linguistic descendants, and I predict that when we sample later (post 5-4kya) individuals we will finally find the West_Asian piece that is missing from the European puzzle.This prediction is now confirmed:
This pattern is also seen in ADMIXTURE analysis (Fig. 2b, SI6), which implies that the Yamnaya have ancestry from populations related to the Caucasus and South Asia that is largely absent in 38 Early or Middle Neolithic farmers but present in all 25 Late Neolithic or Bronze Age individuals. This ancestry appears in Central Europe for the first time in our series with the Corded Ware around 2,500 BCE (SI6, Fig. 2b, Extended Data Fig. 1).I was a little puzzled with the "Ancient North Eurasians" recently proposed as a "third ancestral population" for Europeans: it seemed to be a tertium quid that spread after 5kya, but very different geographically than the "West Asian" component. But:
These results can be explained if the new genetic material that arrived in Germany was a composite of two elements: EHG and a type of Near Eastern ancestry different from that which was introduced by early farmers (also suggested by PCA and ADMIXTURE; Fig. 2, SI5, SI6).So, it seems that there is no contradiction after all and both EHG (which is related to "Ancient North Eurasians") and another type of Near Eastern ancestry (=West_Asian) arrived after 5kya.
1939 strikes back
It is amazing how well this was anticipated by Carleton Coon in 1939. Back then much of West Eurasia was an archaeological/anthropological terra incognita, there was no radiocarbon dating, no DNA, no computers, not even serious multivariate statistics. And yet:
We shall see, in our survey of prehistoric European racial movements, 8 that the Danubian agriculturalists of the Early Neolithic brought a food-producing economy into central Europe from the East. They perpetuated in the new European setting a physical type which was later supplanted in their original home. Several centuries later the Corded people, in the same way, came from southern Russia but there we first find them intermingled with other peoples, and the cul-tural factors which we think of as distinctively Corded are included in a larger cultural equipment. [...] On the basis of the physical evidence as well, it is likely that the Corded people came from somewhere north or east of the Black Sea. The fully Neolithic crania from southern Russia which we have just studied include such a type, also seen in the midst of Sergi's Kurgan aggregation. Until better evidence is produced from elsewhere, we are entitled to consider southern Russia the most likely way station from which the Corded people moved westward.And in 2015:
Our results support a view of European pre-history punctuated by two major migrations: first, the arrival of first farmers during the Early Neolithic from the Near East, and second of Yamnaya pastoralists during the Late Neolithic from the steppe (Extended Data Fig. 5).In 1939:
Linguistically, Indo-European is probably a relatively recent phenomenon, which arose after animals had been tamed and plants cultivated. The latest researches find it to be a derivative of an initially mixed language, whose principal elements were Uralic, called element A, and some undesignated element B which was probably one of the eastern Mediterranean or Caucasic languages. 5 The plants and animals on which the Somewhere in the plains of southern Russia or central Asia, the blending of languages took place which resulted in Indo-European speech. This product in turn spread and split, and was further differentiated by mixture with the languages of peoples upon whom it, in one form or other, was imposed. Some of the present Indo-European languages, in addition to these later accretions from non-Indo-European tongues, contain more of the A element than others, which contain more of the B. The unity of the original " Indo- Europeans," could not have been of long duration, if it was ever complete.In 2015:
These results can be explained if the new genetic material that arrived in Germany was a composite of two elements: EHG and a type of Near Eastern ancestry different from that which was introduced by early farmers (also suggested by PCA and ADMIXTURE; Fig. 2, SI5, SI6). We estimate that these two elements each contributed about half the ancestry each of the Yamnaya (SI6, SI9), explaining why the population turnover inferred using Yamnaya as a source is about twice as high compared to the undiluted EHG.The EHG is still flimsy as it's only two individuals from Karelia and Samara who are very similar to each other. It's hard not to imagine that the hunter-gatherer from Russian Karelia (outside any proposed PIE homeland) would be speaking a similar language as his Samara counterpart. Did they both speak "element A" and was PIE formed when the "southern" steppe hunter-gatherers came into contact with "element B" people from the Caucasus? Short of a time machine, we can never say for sure. This might very well be an answer to the conundrum of Uralic/Proto-Kartvelian borrowings. There is simply no geographical locale in which these two language families neighbor each other: Northwest, Northeast Caucasian speakers and the pesky Greater Caucasus intervene. But, maybe there was no such locale, and these borrowings aren't due to some "PIE people" living adjacent to Uralic and Proto-Karvelian speakers but the "PIE people" being a mix of an element A (EHG) that was (or interacted with) Uralic and another element B (Armenian-like) that was (or interacted with) Proto-Kartvelian.
Urheimat (or not?)
The authors of the current paper are agnostic about the PIE homeland:
We caution that the location of the Proto-Indo-European9,27,29,30 homeland that also gave rise to the Indo-European languages of Asia, as well as the Indo-European languages of southeastern Europe, cannot be determined from the data reported here (SI11). Studying the mixture in the Yamnaya themselves, and understanding the genetic relationships among a broader set of ancient and present-day Indo-European speakers, may lead to new insight about the shared homeland.Whatever the ultimate answer will be, it seems that Coon was right that "The unity of the original " Indo- Europeans," could not have been of long duration, if it was ever complete." If PIE=EHG (as Anthony and Ringe suggest), then "from the crib", PIE got half its ancestry from a non-IE, Near Eastern source. Conversely, if PIE=Near East (as I suggested) then "from the crib", PIE got half of its ancestry from a non-IE, Eastern European source. The "Yamnaya" seems to max out in Norwegians at around half, which means that they are about a quarter Proto-Indo-European genetically, regardless of which theory is right.
These two possibilities (as well as the third one of PIE being neither-nor, but rather a linguistic mixture of the languages of the EHG and Near East) are testable. The Anthony/Ringe version of the steppe hypothesis predicts pre-Yamnaya expansions from the steppe. Whether these happened and what was their makeup can be tested: if they did occur and they did lack "Near Eastern" ancestry, then the steppe hypothesis will be proven. PIE in the Near East, on the other hand, predicts that some PIE languages (certainly the Anatolian ones) will be a "within the Near East" expansion. If such migrations did occur and they lacked "EHG" ancestry, then some variant of the Gamkrelidze/Ivanov model will be proven. Or, the truth might be that everywhere where Indo-Europeans arrive they carry a blend of "West Asian" and "EHG", supporting the third possibility. Time will tell.
In the interim, I am curious about how much Yamnaya ancestry existed in different parts of Europe (all of the post-5kya samples in this study come from Germany, with a couple from Hungary). In northern Europe, all populations seem to have less Yamnaya ancestry than the Corded Ware: there it must have declined. But, modern Hungarians have more than Bronze Age Hungarians: there it must have increased.
Germany and a slice of Hungary is a very narrow window through which to see the whole of Europe and these results must be tested by looking at samples from beyond the "heartland". I do hope that some kind of Moore's law operates in the world of ancient DNA, and in three more years we'll be reading studies about thousands of ancient individuals.
bioRxiv doi: http://dx.doi.org/10.1101/013433
Massive migration from the steppe is a source for Indo-European languages in Europe
Wolfgang Haak , Iosif Lazaridis , Nick Patterson , Nadin Rohland , Swapan Mallick , Bastien Llamas , GuidoBrandt , Susanne Nordenfelt , Eadaoin Harney , Kristin Stewardson , Qiaomei Fu , Alissa Mittnik , Eszter Banffy ,Christos Economou , Michael Francken , Susanne Friederich , Rafael Garrido Pena , Fredrik Hallgren , ValeryKhartanovich , Aleksandr Khokhlov , Michael Kunst , Pavel Kuznetsov , Harald Meller , Oleg Mochalov ,Vayacheslav Moiseyev , Nicole Nicklisch , Sandra L. Pichler , Roberto Risch , Manuel A. Rojo Guerra , ChristinaRoth , Anna Szecsenyi-Nagy , Joachim Wahl , Matthias Meyer , Johannes Krause , Dorcas Brown , DavidAnthony , Alan Cooper , Kurt Werner Alt , David Reich
We generated genome-wide data from 69 Europeans who lived between 8,000-3,000 years ago by enriching ancient DNA libraries for a target set of almost four hundred thousand polymorphisms. Enrichment of these positions decreases the sequencing required for genome-wide ancient DNA analysis by a median of around 250-fold, allowing us to study an order of magnitude more individuals than previous studies and to obtain new insights about the past. We show that the populations of western and far eastern Europe followed opposite trajectories between 8,000-5,000 years ago. At the beginning of the Neolithic period in Europe, ~8,000-7,000 years ago, closely related groups of early farmers appeared in Germany, Hungary, and Spain, different from indigenous hunter-gatherers, whereas Russia was inhabited by a distinctive population of hunter-gatherers with high affinity to a ~24,000 year old Siberian6. By ~6,000-5,000 years ago, a resurgence of hunter-gatherer ancestry had occurred throughout much of Europe, but in Russia, the Yamnaya steppe herders of this time were descended not only from the preceding eastern European hunter-gatherers, but from a population of Near Eastern ancestry. Western and Eastern Europe came into contact ~4,500 years ago, as the Late Neolithic Corded Ware people from Germany traced ~3/4 of their ancestry to the Yamnaya, documenting a massive migration into the heartland of Europe from its eastern periphery. This steppe ancestry persisted in all sampled central Europeans until at least ~3,000 years ago, and is ubiquitous in present-day Europeans. These results provide support for the theory of a steppe origin of at least some of the Indo-European languages of Europe.
Link
January 14, 2015
Y chromosome super-fathers

The most interesting one is DC2 which was also found in south Siberian Kurgans, belongs to haplogroup R1a1 and is given an age of 3,284 years by the authors (with some almost impossibly wide confidence intervals). Based on its distribution, and if a Bronze Age date is right, it is difficult to see in it anything other than a patrilineage that was present in Proto-Indo-Iranians.

European Journal of Human Genetics advance online publication 14 January 2015; doi: 10.1038/ejhg.2014.285
Y-chromosome descent clusters and male differential reproductive success: young lineage expansions dominate Asian pastoral nomadic populations
Patricia Balaresque et al.
High-frequency microsatellite haplotypes of the male-specific Y-chromosome can signal past episodes of high reproductive success of particular men and their patrilineal descendants. Previously, two examples of such successful Y-lineages have been described in Asia, both associated with Altaic-speaking pastoral nomadic societies, and putatively linked to dynasties descending, respectively, from Genghis Khan and Giocangga. Here we surveyed a total of 5321 Y-chromosomes from 127 Asian populations, including novel Y-SNP and microsatellite data on 461 Central Asian males, to ask whether additional lineage expansions could be identified. Based on the most frequent eight-microsatellite haplotypes, we objectively defined 11 descent clusters (DCs), each within a specific haplogroup, that represent likely past instances of high male reproductive success, including the two previously identified cases. Analysis of the geographical patterns and ages of these DCs and their associated cultural characteristics showed that the most successful lineages are found both among sedentary agriculturalists and pastoral nomads, and expanded between 2100 BCE and 1100 CE. However, those with recent origins in the historical period are almost exclusively found in Altaic-speaking pastoral nomadic populations, which may reflect a shift in political organisation in pastoralist economies and a greater ease of transmission of Y-chromosomes through time and space facilitated by the use of horses.
Link
January 01, 2015
Y-chromosome tree bursts to leaf (Hallast, Batini, Zadik et al. 2014)
This is an extremely important study of Y-chromosome variation, the most intriguing part of which are the copious references to a yet to come manuscript:
The Y-Chromosome Tree Bursts into Leaf: 13,000 High-Confidence SNPs Covering the Majority of Known Clades
Pille Hallast, Chiara Batini, Daniel Zadik et al.
Many studies of human populations have used the male-specific region of the Y chromosome (MSY) as a marker, but MSY sequence variants have traditionally been subject to ascertainment bias. Also, dating of haplogroups has relied on Y-specific short tandem repeats (STRs), involving problems of mutation rate choice, and possible long-term mutation saturation. Next-generation sequencing can ascertain single nucleotide polymorphisms (SNPs) in an unbiased way, leading to phylogenies in which branch-lengths are proportional to time, and allowing the times-to-most-recent-common-ancestor (TMRCAs) of nodes to be estimated directly. Here we describe the sequencing of 3.7 Mb of MSY in each of 448 human males at a mean coverage of 51×, yielding 13,261 high-confidence SNPs, 65.9% of which are previously unreported. The resulting phylogeny covers the majority of the known clades, provides date estimates of nodes, and constitutes a robust evolutionary framework for analyzing the history of other classes of mutation. Different clades within the tree show subtle but significant differences in branch lengths to the root. We also apply a set of 23 Y-STRs to the same samples, allowing SNP- and STR-based diversity and TMRCA estimates to be systematically compared. Ongoing purifying selection is suggested by our analysis of the phylogenetic distribution of nonsynonymous variants in 15 MSY single-copy genes.
Link
Elsewhere (Batini C, Hallast P, Zadik D, Maisano Delser P, Benazzo A, Ghirotto S, Arroyo-Pardo E, Cavalleri GL, de Knijff P, Dupuy BM, Eriksen H, King TE, López de Munain A, López-Parra AM, Milasin J, Novelletto A, Pamjav H, Sajantila A, Tolun A, Winney B and Jobling MA, submitted.) we have described an NGS-based MSY phylogeny based on 5,996 SNPs ascertained in 334 human Y chromosomes comprising 17 population samples from Europe and the Near East, focused on illuminating the origins and histories of European patrilineages.Anyway, the current paper is openly available, so do read it if you haven't already. Of interest to long-time readers of this blog is this bit:
Generally, the STRs perform poorly, giving a wide variety of TMRCAs for nodes with similar SNP-based dates, and correlation coefficients consistently below 0.6. Considering the variables described above: 1) ASD generally outperforms rho, and choice of rooting method (ancestral or modal) makes little difference. For rho, rooting through the ancestral haplotype performs much worse than through the modal haplotype; 2) removal of RM-YSTRs, and STRs showing repeat array complexity, does not have a major influence on relationships between SNP- and STR-based estimates of TMRCA, and the effects depend upon how the root is specified; and 3) the evolutionary STR mutation rate consistently overestimates, and the pedigree rate underestimates, the TMRCAs of nodes (fig. 4a). As expected, the pedigree mutation rate performs better for young nodes (less than 10 ka; supplementary table S6, Supplementary Material online), whereas the evolutionary rate performs better for older nodes.Mol Biol Evol (2014) doi: 10.1093/molbev/msu327
The Y-Chromosome Tree Bursts into Leaf: 13,000 High-Confidence SNPs Covering the Majority of Known Clades
Pille Hallast, Chiara Batini, Daniel Zadik et al.
Many studies of human populations have used the male-specific region of the Y chromosome (MSY) as a marker, but MSY sequence variants have traditionally been subject to ascertainment bias. Also, dating of haplogroups has relied on Y-specific short tandem repeats (STRs), involving problems of mutation rate choice, and possible long-term mutation saturation. Next-generation sequencing can ascertain single nucleotide polymorphisms (SNPs) in an unbiased way, leading to phylogenies in which branch-lengths are proportional to time, and allowing the times-to-most-recent-common-ancestor (TMRCAs) of nodes to be estimated directly. Here we describe the sequencing of 3.7 Mb of MSY in each of 448 human males at a mean coverage of 51×, yielding 13,261 high-confidence SNPs, 65.9% of which are previously unreported. The resulting phylogeny covers the majority of the known clades, provides date estimates of nodes, and constitutes a robust evolutionary framework for analyzing the history of other classes of mutation. Different clades within the tree show subtle but significant differences in branch lengths to the root. We also apply a set of 23 Y-STRs to the same samples, allowing SNP- and STR-based diversity and TMRCA estimates to be systematically compared. Ongoing purifying selection is suggested by our analysis of the phylogenetic distribution of nonsynonymous variants in 15 MSY single-copy genes.
Link
Y Chromosome of Aisin Gioro: C3b2b1
Apart from the historical interest, this study might be useful to further calibrate the Y-chromosome molecular clock. The Y-SNP mutation rate was previously calibrated with a Chinese pedigree that went down to ~1800AD, and this is potentially much deeper.
arXiv:1412.6274 [q-bio.PE]
Y Chromosome of Aisin Gioro, the Imperial House of Qing Dynasty
Shi Yan, Harumasa Tachibana, Lan-Hai Wei, Ge Yu, Shao-Qing Wen, Chuan-Chao Wang
(Submitted on 19 Dec 2014) House of Aisin Gioro is the imperial family of the last dynasty in Chinese history - Qing Dynasty (1644 - 1911). Aisin Gioro family originated from Jurchen tribes and developed the Manchu people before they conquered China. By investigating the Y chromosomal short tandem repeats (STRs) of 7 modern male individuals who claim belonging to Aisin Gioro family (in which 3 have full records of pedigree), we found that 3 of them (in which 2 keep full pedigree, whose most recent common ancestor is Nurgaci) shows very close relationship (1 - 2 steps of difference in 17 STR) and the haplotype is rare. We therefore conclude that this haplotype is the Y chromosome of the House of Aisin Gioro. Further tests of single nucleotide polymorphisms (SNPs) indicates that they belong to Haplogroup C3b2b1*-M401(xF5483), although their Y-STR results are distant to the "star cluster", which also belongs to the same haplogroup. This study forms the base for the pedigree research of the imperial family of Qing Dynasty by means of genetics.
Link
arXiv:1412.6274 [q-bio.PE]
Y Chromosome of Aisin Gioro, the Imperial House of Qing Dynasty
Shi Yan, Harumasa Tachibana, Lan-Hai Wei, Ge Yu, Shao-Qing Wen, Chuan-Chao Wang
(Submitted on 19 Dec 2014) House of Aisin Gioro is the imperial family of the last dynasty in Chinese history - Qing Dynasty (1644 - 1911). Aisin Gioro family originated from Jurchen tribes and developed the Manchu people before they conquered China. By investigating the Y chromosomal short tandem repeats (STRs) of 7 modern male individuals who claim belonging to Aisin Gioro family (in which 3 have full records of pedigree), we found that 3 of them (in which 2 keep full pedigree, whose most recent common ancestor is Nurgaci) shows very close relationship (1 - 2 steps of difference in 17 STR) and the haplotype is rare. We therefore conclude that this haplotype is the Y chromosome of the House of Aisin Gioro. Further tests of single nucleotide polymorphisms (SNPs) indicates that they belong to Haplogroup C3b2b1*-M401(xF5483), although their Y-STR results are distant to the "star cluster", which also belongs to the same haplogroup. This study forms the base for the pedigree research of the imperial family of Qing Dynasty by means of genetics.
Link
December 02, 2014
Remains of Richard III identified
From the paper:
Four of the modern relatives were found to belong to Y-haplogroup R1b-U152 (x L2, Z36, Z56, M160, M126 and Z192)13, 14 with STR haplotypes being consistent with them comprising a single patrilinear group. One individual (Somerset 3) was found to belong to haplogroup I-M170 (x M253, M223) and therefore could not be a patrilinear relative of the other four within the time span considered, indicating that a false-paternity event had occurred within the last four generations.
...
In contrast to the Y-haplotypes of the putative modern relatives, Skeleton 1 belongs to haplogroup G-P287, with a corresponding Y-STR haplotype. Thus, the putative modern patrilinear relatives of Richard III are not genetically related to Skeleton 1 through the male line over the time period considered. However, this is not surprising, given an estimated average false-paternity rate of ~1–2% (refs 12, 17, 18). The putative modern relatives and Richard III are related through a male relative (Edward III) four generations up from Richard III (Fig. 1a and Supplementary Fig. 2), and a false-paternity event could have happened in any of the 19 generations separating Richard III and the 5th Duke of Beaufort, on either branch of the genealogy descending from Edward III. Indeed, even with a conservative false-paternity rate18 (see Supplementary Methods) the chance of a false-paternity occuring in this number of generations is 16%.
Nature Communications 5, Article number: 5631 doi:10.1038/ncomms6631
Identification of the remains of King Richard III
Turi E. King et al.
Abstract
In 2012, a skeleton was excavated at the presumed site of the Grey Friars friary in Leicester, the last-known resting place of King Richard III. Archaeological, osteological and radiocarbon dating data were consistent with these being his remains. Here we report DNA analyses of both the skeletal remains and living relatives of Richard III. We find a perfect mitochondrial DNA match between the sequence obtained from the remains and one living relative, and a single-base substitution when compared with a second relative. Y-chromosome haplotypes from male-line relatives and the remains do not match, which could be attributed to a false-paternity event occurring in any of the intervening generations. DNA-predicted hair and eye colour are consistent with Richard’s appearance in an early portrait. We calculate likelihood ratios for the non-genetic and genetic data separately, and combined, and conclude that the evidence for the remains being those of Richard III is overwhelming.
Link
Subscribe to:
Posts (Atom)