The American Journal of Human Genetics, volume 79 (2006)
A Geographically Explicit Genetic Model of Worldwide Human-Settlement History
Hua Liu et al.
Currently available genetic and archaeological evidence is generally interpreted as supportive of a recent single origin of modern humans in East Africa. However, this is where the near consensus on human settlement history ends, and considerable uncertainty clouds any more detailed aspect of human colonization history. Here, we present a dynamic genetic model of human settlement history coupled with explicit geographical distances from East Africa, the likely origin of modern humans. We search for the best-supported parameter space by fitting our analytical prediction to genetic data that are based on 52 human populations analyzed at 783 autosomal microsatellite markers. This framework allows us to jointly estimate the key parameters of the expansion of modern humans. Our best estimates suggest an initial expansion of modern humans ~56,000 years ago from a small founding population of ~1,000 effective individuals. Our model further points to high growth rates in newly colonized habitats. The general fit of the model with the data is excellent. This suggests that coupling analytical genetic models with explicit demography and geography provides a powerful tool for making inferences on human-settlement history.
Link

May 31, 2006
May 26, 2006
Absence of father or unhappy homes => ugly daughters
UPDATE
Many theories proposed in the comments section below.. Here is my take on a possible explanation. It is expected that a woman's ability to retain her husband and create a happy home is positively correlated to her attractiveness [Note that I am not saying that this is the only factor]. Males are more ready to leave their spouses if they are not attractive, e.g., if they find, or hope to find, another woman that is more attractive. If they decide to stay in the marriage, then the knowledge that their wives are unattractive, and especially the comparison with other women (of one's friends or celebrities) will be a constant source of disappointment, leading to marital friction. Hence, daughters in homes with absent fathers or unhappy homes are less attractive because their mothers are less attractive.
I should say that the authors do propose alternative explanations in their paper, although their data cannot be used to differentiate between them:
ISSN: 0962-8452 (Paper) 1471-2954 (Online)
Facial and bodily correlates of family background
Lynda G. Boothroyd et al.
Abstract
It has been suggested that absence of the father during early childhood has long-reaching effects on reproductive strategy and development of offspring. This paper reports two studies designed to investigate the physical characteristics of daughters associated with father absence. Study 1 used a facial averaging method to produce composite images of faces of women whose parents separated during their childhood (who were 'father absent'), women whose parents remained together, but had poor quality relationships and women whose parents were together and had good quality relationships. Images were then rated by male and female judges. Father absence and poor parental relationships were associated with apparent facial masculinity and reduced attractiveness in daughters. Poor parental relationships were also associated with reduced apparent health. Study 2 compared family background with body measurements and found that father absence or a poor quality relationship between parents were associated with body masculinity (high waist-to-hip ratio) and increased weight-for-height and adiposity. These results highlight the possibility of physical masculinization being associated with purported father absence 'effects'.
Link
Many theories proposed in the comments section below.. Here is my take on a possible explanation. It is expected that a woman's ability to retain her husband and create a happy home is positively correlated to her attractiveness [Note that I am not saying that this is the only factor]. Males are more ready to leave their spouses if they are not attractive, e.g., if they find, or hope to find, another woman that is more attractive. If they decide to stay in the marriage, then the knowledge that their wives are unattractive, and especially the comparison with other women (of one's friends or celebrities) will be a constant source of disappointment, leading to marital friction. Hence, daughters in homes with absent fathers or unhappy homes are less attractive because their mothers are less attractive.
I should say that the authors do propose alternative explanations in their paper, although their data cannot be used to differentiate between them:
It is important to note that this study cannot distinguish between the possible environmental and hereditary influences underlying the relationships between family background and physical development. There are several ways in which health of offspring might be linked to parental relationship. Family stress may negatively impact on offspring health via the immunosuppressant effects of cortisol or high-stress families may have less healthy lifestyles. Alternatively, heritable parental health may influence both offspring health and parental marital relations. Finally, less healthy children may be a catalyst for difficult relationships between parents.Proceedings of The Royal Society B
ISSN: 0962-8452 (Paper) 1471-2954 (Online)
Facial and bodily correlates of family background
Lynda G. Boothroyd et al.
Abstract
It has been suggested that absence of the father during early childhood has long-reaching effects on reproductive strategy and development of offspring. This paper reports two studies designed to investigate the physical characteristics of daughters associated with father absence. Study 1 used a facial averaging method to produce composite images of faces of women whose parents separated during their childhood (who were 'father absent'), women whose parents remained together, but had poor quality relationships and women whose parents were together and had good quality relationships. Images were then rated by male and female judges. Father absence and poor parental relationships were associated with apparent facial masculinity and reduced attractiveness in daughters. Poor parental relationships were also associated with reduced apparent health. Study 2 compared family background with body measurements and found that father absence or a poor quality relationship between parents were associated with body masculinity (high waist-to-hip ratio) and increased weight-for-height and adiposity. These results highlight the possibility of physical masculinization being associated with purported father absence 'effects'.
Link
Barbujani and Chikhi on genetic contribution of Neolithic
The recent study of Neolithic mtDNA from Central European proto-farmers was interpreted by the authors of the study as evidence for limited contribution of the Neolithic in the modern European gene pool. From the cited commentary:
Heredity advance online publication 24 May 2006; doi: 10.1038/sj.hdy.6800852
Population genetics: DNAs from the European Neolithic
G Barbujani and L Chikhi
Link
As noted above, a major part of the controversy has revolved around the age of haplogroups. Since these ages were used by Richards and collaborators to identify 'palaeolithic' and 'neolithic' components of the modern gene pool, one would expect Neolithic specimens to only yield 'neolithic', haplogroup J, sequences. The population genetics prediction, however, is that 'Neolithic' people should have both types of haplogroups. Interestingly, Haak et al (2005) found only one sequence belonging to haplogroup J, with six sequences belonging to the currently very rare N1a haplogroup, and 17 to haplogroups that were termed 'Paleolithic' (Richards et al, 2000), such as H, V and K. Surprisingly, this information was not used by the authors, even though it demonstrates that ages of molecules cannot be equated with ages of populations, a point made some time ago by supporters of the demic diffusion model (Barbujani et al, 1998). Population genetics theory teaches us that migrating people carry alleles and haplogroups in their genome originating from mutations that occurred before, sometimes long before, the migratory movement started, and inferring from the former the date of the latter is never straightforward. It might be legitimate (although, we think, misleading) to term haplotypes derived from mutations <10 000 years old as 'Neolithic', but the frequency of those haplotypes has little to do with the Neolithic contribution to the European gene pool.
Heredity advance online publication 24 May 2006; doi: 10.1038/sj.hdy.6800852
Population genetics: DNAs from the European Neolithic
G Barbujani and L Chikhi
Link
Fine resolution study of mtDNA haplogroup H
Electrophoresis. 2006 May 24; [Epub ahead of print]
Dissection of mitochondrial superhaplogroup H using coding region SNPs.
Brandstatter A, Salas A, Niederstatter H, Gassner C, Carracedo A, Parson W.
Haplogroup H (hg H) includes about 40-50% of the West Eurasian mitochondrial DNA (mtDNA) samples investigated so far. In order to enhance discrimination within this haplogroup we selected 45 coding region SNPs that allow to ascribe samples to the main phylogenetic branches of super hg HV (that embraces hg H) and, in particular, to H sublineages with a much finer resolution than previous studies. SNP selection was carried out using the most up-to-date available literature on population and forensic genetics and extended by means of phylogenetic analysis of complete or coding region genomes (<430) and control region sequences. A meticulous inspection of the H phylogeny led us to the observation of various but uncharacterized subclades of hg H. The selected SNPs were amplified in two PCR-multiplex reactions and subsequently targeted in three single-base extension multiplex reactions. A total of 2214 West Eurasian samples were screened for hg H specific loci 2706 and 7028, of which 859 fell in hg H and were further subjected to subhaplogroup typing. We observed 35 different subhaplogroups in total, 33 of which were found at frequencies below 5%. This assay can be used as a prescreening tool in forensic casework for rapid discrimination between divergent lineages (very effective for high-volume crime cases) or as discriminatory assay, when identical hg H haplotypes were obtained by control region sequencing.
Link
Dissection of mitochondrial superhaplogroup H using coding region SNPs.
Brandstatter A, Salas A, Niederstatter H, Gassner C, Carracedo A, Parson W.
Haplogroup H (hg H) includes about 40-50% of the West Eurasian mitochondrial DNA (mtDNA) samples investigated so far. In order to enhance discrimination within this haplogroup we selected 45 coding region SNPs that allow to ascribe samples to the main phylogenetic branches of super hg HV (that embraces hg H) and, in particular, to H sublineages with a much finer resolution than previous studies. SNP selection was carried out using the most up-to-date available literature on population and forensic genetics and extended by means of phylogenetic analysis of complete or coding region genomes (<430) and control region sequences. A meticulous inspection of the H phylogeny led us to the observation of various but uncharacterized subclades of hg H. The selected SNPs were amplified in two PCR-multiplex reactions and subsequently targeted in three single-base extension multiplex reactions. A total of 2214 West Eurasian samples were screened for hg H specific loci 2706 and 7028, of which 859 fell in hg H and were further subjected to subhaplogroup typing. We observed 35 different subhaplogroups in total, 33 of which were found at frequencies below 5%. This assay can be used as a prescreening tool in forensic casework for rapid discrimination between divergent lineages (very effective for high-volume crime cases) or as discriminatory assay, when identical hg H haplotypes were obtained by control region sequencing.
Link
May 24, 2006
Y chromosomes of Sweden
Haplogroup frequencies:
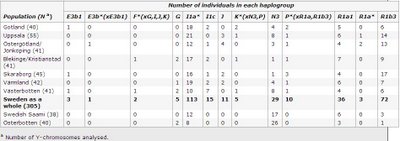
European Journal of Human Genetics (advance online publication)
Y-chromosome diversity in Sweden – A long-time perspective
Andreas O Karlsson et al.
Abstract
Sixteen Y-chromosomal binary markers and nine Y-chromosome short tandem repeats were analyzed in a total of 383 unrelated males from seven different Swedish regions, one Finnish region and a Swedish Saami population in order to address questions about the origin and genetic structure of the present day population in Sweden. Haplogroup I1a* was found to be the most common haplogroup in Sweden and accounted, together with haplogroups R1b3, R1a1 and N3, for over 80% of the male lineages. Within Sweden, a minor stratification was found in which the northern region Västerbotten differed significantly (P<0.05) from the other Swedish regions. A flow of N3 chromosomes into Västerbotten mainly from Saami and Finnish populations could be one explanation for this stratification. However, the demographic history of Västerbotten involving a significant male absence during the 17th Century may also have had a large impact. Immigration of young men from elsewhere to Värmland at the same time, can be responsible for a similar deviation with I1a* haplotypes. Y chromosomes within haplogroup R1b3 were found to have the highest STR variation among all haplogroups and could thus be considered to be one of the earliest major male lineages present in Sweden. Regional haplotype variation, within R1b3, also showed a difference between two regions in the south of Sweden. This can also be traced from historical time and is visible in archaeological material. Overall this Y chromosome study provides interesting information about the genetic patterns and demographic events in the Swedish population.
Link

European Journal of Human Genetics (advance online publication)
Y-chromosome diversity in Sweden – A long-time perspective
Andreas O Karlsson et al.
Abstract
Sixteen Y-chromosomal binary markers and nine Y-chromosome short tandem repeats were analyzed in a total of 383 unrelated males from seven different Swedish regions, one Finnish region and a Swedish Saami population in order to address questions about the origin and genetic structure of the present day population in Sweden. Haplogroup I1a* was found to be the most common haplogroup in Sweden and accounted, together with haplogroups R1b3, R1a1 and N3, for over 80% of the male lineages. Within Sweden, a minor stratification was found in which the northern region Västerbotten differed significantly (P<0.05) from the other Swedish regions. A flow of N3 chromosomes into Västerbotten mainly from Saami and Finnish populations could be one explanation for this stratification. However, the demographic history of Västerbotten involving a significant male absence during the 17th Century may also have had a large impact. Immigration of young men from elsewhere to Värmland at the same time, can be responsible for a similar deviation with I1a* haplotypes. Y chromosomes within haplogroup R1b3 were found to have the highest STR variation among all haplogroups and could thus be considered to be one of the earliest major male lineages present in Sweden. Regional haplotype variation, within R1b3, also showed a difference between two regions in the south of Sweden. This can also be traced from historical time and is visible in archaeological material. Overall this Y chromosome study provides interesting information about the genetic patterns and demographic events in the Swedish population.
Link
May 23, 2006
Updating the East Asian mtDNA phylogeny
Hum Mol Genet. 2006 May 19; [Epub ahead of print]
Updating the East Asian mtDNA Phylogeny: A Prerequisite for the Identification of Pathogenic Mutations.
Kong QP, Bandelt HJ, Sun C, Yao YG, Salas A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, Torroni A, Zhang YP.
Knowledge about the world phylogeny of human mitochondrial DNA (mtDNA) is essential not only for evaluating the pathogenic role of specific mtDNA mutations but also for performing reliable association studies between mtDNA haplogroups and complex disorders. In the past few years, the main features of the East Asian portion of the mtDNA phylogeny have been determined based on complete sequencing efforts, but representatives of several basal lineages were still lacking. Moreover, some recently published complete mtDNA sequences did apparently not fit into the known phylogenetic tree and conflicted with the established nomenclature. To refine the East Asian mtDNA tree and resolve data conflicts, we first completely sequenced 20 carefully selected mtDNAs - likely representatives of novel subhaplogroups - and then, in order to distinguish diagnostic mutations of novel haplogroups from private variants, we applied a "motif-search" procedure to a large sample collection. The novel information was incorporated into an updated East Asian mtDNA tree encompassing more than 1000 (near-) complete mtDNA sequences. A reassessment of the mtDNA data from a series of disease studies testified to the usefulness of such a refined mtDNA tree in evaluating the pathogenicity of mtDNA mutations. In particular, the claimed pathogenic role of mutations G3316A, T3394C, A4833G, and G15497A appears to be most questionable as those initial claims were derived from anecdotal findings rather than association studies. Following a guideline based on the phylogenetic knowledge as proposed here could help avoiding similar problems in the future.
Link
Updating the East Asian mtDNA Phylogeny: A Prerequisite for the Identification of Pathogenic Mutations.
Kong QP, Bandelt HJ, Sun C, Yao YG, Salas A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, Torroni A, Zhang YP.
Knowledge about the world phylogeny of human mitochondrial DNA (mtDNA) is essential not only for evaluating the pathogenic role of specific mtDNA mutations but also for performing reliable association studies between mtDNA haplogroups and complex disorders. In the past few years, the main features of the East Asian portion of the mtDNA phylogeny have been determined based on complete sequencing efforts, but representatives of several basal lineages were still lacking. Moreover, some recently published complete mtDNA sequences did apparently not fit into the known phylogenetic tree and conflicted with the established nomenclature. To refine the East Asian mtDNA tree and resolve data conflicts, we first completely sequenced 20 carefully selected mtDNAs - likely representatives of novel subhaplogroups - and then, in order to distinguish diagnostic mutations of novel haplogroups from private variants, we applied a "motif-search" procedure to a large sample collection. The novel information was incorporated into an updated East Asian mtDNA tree encompassing more than 1000 (near-) complete mtDNA sequences. A reassessment of the mtDNA data from a series of disease studies testified to the usefulness of such a refined mtDNA tree in evaluating the pathogenicity of mtDNA mutations. In particular, the claimed pathogenic role of mutations G3316A, T3394C, A4833G, and G15497A appears to be most questionable as those initial claims were derived from anecdotal findings rather than association studies. Following a guideline based on the phylogenetic knowledge as proposed here could help avoiding similar problems in the future.
Link
May 20, 2006
mtDNA variation in Jewish populations
Haplogroup frequencies from the paper:
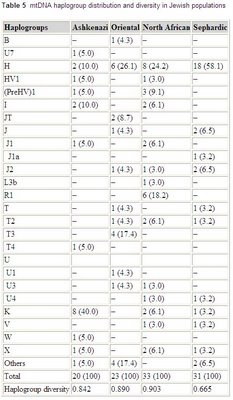
Int J Legal Med. 2006 May 18; [Epub ahead of print]
Mitochondrial DNA sequence variation in Jewish populations.
Picornell A, Gimenez P, Castro JA, Ramon MM.
Sequence analysis of HVRI and HVRII mitochondrial DNA was carried out on 107 Jewish samples from Ashkenazi, Oriental, North African, and Sephardic origins. Control region sequences were assigned to a haplogroup by means of the analysis of the RFLP motif -7025 AluI or by using sequence motifs. A total of 88 different haplotypes were observed with a lower incidence of unique haplotypes (68.2%) than in other populations. Four individuals with one position of sequence heteroplasmy at nucleotides 16093, 16134, 16169, and 235, respectively, were detected. The mean pairwise difference in the Jewish population was 9.7 nucleotides. The gene diversity was 0.996, and the random match probability was 1.3%. When the data were compared with the autosomal and Y-chromosome markers previously studied in these populations, sex-specific differences could be observed in the Jewish populations. This fact must be taken into account for choosing suitable databases to correctly weigh the value of the evidence of a mtDNA and/or Y profile match.
Link
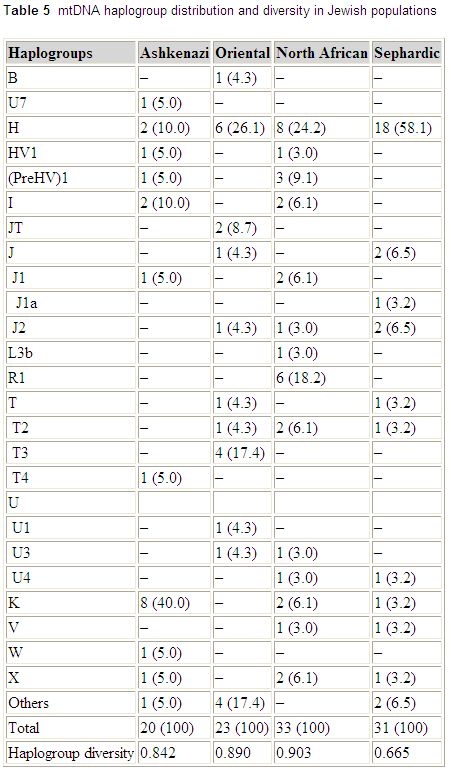
Int J Legal Med. 2006 May 18; [Epub ahead of print]
Mitochondrial DNA sequence variation in Jewish populations.
Picornell A, Gimenez P, Castro JA, Ramon MM.
Sequence analysis of HVRI and HVRII mitochondrial DNA was carried out on 107 Jewish samples from Ashkenazi, Oriental, North African, and Sephardic origins. Control region sequences were assigned to a haplogroup by means of the analysis of the RFLP motif -7025 AluI or by using sequence motifs. A total of 88 different haplotypes were observed with a lower incidence of unique haplotypes (68.2%) than in other populations. Four individuals with one position of sequence heteroplasmy at nucleotides 16093, 16134, 16169, and 235, respectively, were detected. The mean pairwise difference in the Jewish population was 9.7 nucleotides. The gene diversity was 0.996, and the random match probability was 1.3%. When the data were compared with the autosomal and Y-chromosome markers previously studied in these populations, sex-specific differences could be observed in the Jewish populations. This fact must be taken into account for choosing suitable databases to correctly weigh the value of the evidence of a mtDNA and/or Y profile match.
Link
May 19, 2006
Hobbit Sparks Duel of the Anthropologists
This announcement from the Field Museum is one side of the story:
Scientists scuttle claims that 'Hobbit' fossil from Flores, Indonesia, is a new hominid (excerpt):
BTW, here is a picture of a human microcephalic, and another (bottom).
And, finally the abstracts of the two papers.
Comment on "The Brain of LB1, Homo floresiensis"
R. D. Martin, A. M. MacLarnon, J. L. Phillips, L. Dussubieux, P. R. Williams, and W. B. Dobyns
Science 19 May 2006: 999.
Endocast analysis of the brain Homo floresiensis by Falk et al. (Reports, 8 April 2005, p. 242) implies that the hominid is an insular dwarf derived from H. erectus, but its tiny cranial capacity cannot result from normal dwarfing. Consideration of more appropriate microcephalic syndromes and specimens supports the hypothesis of modern human microcephaly.
Link
Response to Comment on "The Brain of LB1, Homo floresiensis"
Dean Falk, Charles Hildebolt, Kirk Smith, M. J. Morwood, Thomas Sutikna, Jatmiko, E. Wayhu Saptomo, Barry Brunsden, and Fred Prior
Science 19 May 2006: 999.
Martin et al. claim that they have two endocasts from microcephalics that appear similar to that of LB1, Homo floresiensis. However, the line drawings they present as evidence lack details about the transverse sinuses, cerebellum, and cerebral poles. Comparative measurements, actual photographs, and sketches that identify key features are needed to draw meaningful conclusions about Martin et al.'s assertions.
Link
Scientists scuttle claims that 'Hobbit' fossil from Flores, Indonesia, is a new hominid (excerpt):
New evidence highlights failure to respect good scientific practiceMore on the controversy from the Washington Post (excerpt):
CHICAGO--When scientists found 18,000-year-old bones of a small, humanlike creature on the Indonesian island of Flores in 2003, they concluded that the bones represented a new species in the human family tree that they named Homo floresiensis. Their interpretation was widely accepted by the scientific community and heralded by the popular press around the world. Because of its very short stature, H. floresiensis was soon dubbed the "Hobbit."
Increasingly, however, this controversial conclusion is being questioned. In a Technical Comment to be published in the May 19, 2006, issue of Science magazine, scientists led by Robert D. Martin, PhD, Field Museum Provost and world-class primatologist, say that the bones in question do not represent a new species at all. A far more likely explanation is that the bones belonged to a modern human who suffered from microcephaly, a pathological condition that causes small brain size, often associated with short stature.
A research team led by primatologist Robert D. Martin, provost of Chicago's Field Museum, argues that no human ancestor could reach a weight of 64 pounds with a brain size of only 23.2 cubic inches and be able to make sophisticated tools like those found with the Hobbit remains.Many scientific controversies about paleoanthropological remains are quickly resolved if new facts emerge, but this is a battle which doesn't seem to be going away any time soon, with both sides sticking to their guns.
The Martin team said the Hobbit must have been a modern human with microcephaly -- a condition, usually genetic, in which the brain fails to grow to normal size. "This brain is too small for any explanation besides pathology," Martin said in a telephone interview.
In a rebuttal to the Martin group, a second team led by Florida State University paleoanthropologist Dean Falk defended their earlier research contending that the Flores skull was nothing like that of a microcephalic, and that the remains most likely represent a previously unknown species.
"We are just finishing a big study on microcephalics that confirms our earlier observations," Falk said in a telephone interview. While microcephalic brains shrink with age, causing the inside of the skull to smooth out, the Flores skull is highly convoluted, reflecting the imprint of a fully expanded, fully functioning brain, she said.
BTW, here is a picture of a human microcephalic, and another (bottom).
And, finally the abstracts of the two papers.
Comment on "The Brain of LB1, Homo floresiensis"
R. D. Martin, A. M. MacLarnon, J. L. Phillips, L. Dussubieux, P. R. Williams, and W. B. Dobyns
Science 19 May 2006: 999.
Endocast analysis of the brain Homo floresiensis by Falk et al. (Reports, 8 April 2005, p. 242) implies that the hominid is an insular dwarf derived from H. erectus, but its tiny cranial capacity cannot result from normal dwarfing. Consideration of more appropriate microcephalic syndromes and specimens supports the hypothesis of modern human microcephaly.
Link
Response to Comment on "The Brain of LB1, Homo floresiensis"
Dean Falk, Charles Hildebolt, Kirk Smith, M. J. Morwood, Thomas Sutikna, Jatmiko, E. Wayhu Saptomo, Barry Brunsden, and Fred Prior
Science 19 May 2006: 999.
Martin et al. claim that they have two endocasts from microcephalics that appear similar to that of LB1, Homo floresiensis. However, the line drawings they present as evidence lack details about the transverse sinuses, cerebellum, and cerebral poles. Comparative measurements, actual photographs, and sketches that identify key features are needed to draw meaningful conclusions about Martin et al.'s assertions.
Link
May 17, 2006
Microcephalin and ASPM do not Account for Brain Size Variability
Related to these recent papers, a new study on Microcephalin and ASPM, the "brain genes" under recent selection.
Human Molecular Genetics (Advance Access published online on May 10, 2006)
Normal Variants of Microcephalin and ASPM Do Not Account for Brain Size Variability
Roger P. Woods et al.
Abstract
Normal human brain volume is heritable. The genes responsible for variation in brain volume are not known. Microcephalin (MCPH1) and ASPM (abnormal spindle-like microcephaly associated) have been proposed as candidate genes since mutations in both genes are associated with microcephaly and common variants of each gene are apparently under strong positive selective pressure. In 120 normal subjects, we genotyped these variants and measured brain volumes using magnetic resonance imaging. We found no evidence that the selected alleles were associated with increases or decreases in brain volume. This result suggests that the selective pressure on these genes may be related to subtle neurobiological effects or to their expression outside the brain.
Link
Human Molecular Genetics (Advance Access published online on May 10, 2006)
Normal Variants of Microcephalin and ASPM Do Not Account for Brain Size Variability
Roger P. Woods et al.
Abstract
Normal human brain volume is heritable. The genes responsible for variation in brain volume are not known. Microcephalin (MCPH1) and ASPM (abnormal spindle-like microcephaly associated) have been proposed as candidate genes since mutations in both genes are associated with microcephaly and common variants of each gene are apparently under strong positive selective pressure. In 120 normal subjects, we genotyped these variants and measured brain volumes using magnetic resonance imaging. We found no evidence that the selected alleles were associated with increases or decreases in brain volume. This result suggests that the selective pressure on these genes may be related to subtle neurobiological effects or to their expression outside the brain.
Link
Mother's mitochondrial curse
Trends Ecol Evol. 2004 May;19(5):238-44.
Mother's curse: the effect of mtDNA on individual fitness and population viability.
Gemmell NJ, Metcalf VJ, Allendorf FW.
The mitochondrial genome is considered generally to be an innocent bystander in adaptive evolution; however, there is increasing evidence that mitochondrial DNA (mtDNA) is an important contributor to viability and fecundity. Some of this evidence is now well documented, with mtDNA mutations having been shown to play a causal role in degenerative diseases, ageing, and cancer. However, most research on mtDNA has ignored the possibility that other instances exist where mtDNA mutations could have profound fitness consequences. Recent work in humans and other species now indicates that mtDNA mutations play an important role in sperm function, male fertility, and male fitness. Ironically, deleterious mtDNA mutations that affect only males, such as those that impair sperm function, will not be subject to natural selection because mitochondria are generally maternally inherited and could reach high frequencies in populations if the mutations are not disadvantageous in females. Here, we review how such mtDNA mutations might affect the viability of natural populations. We consider factors that increase or decrease the strength of the effect of mtDNA mutations on population viability and discuss what mechanisms exist to mitigate deleterious mtDNA effects.
Link
Mother's curse: the effect of mtDNA on individual fitness and population viability.
Gemmell NJ, Metcalf VJ, Allendorf FW.
The mitochondrial genome is considered generally to be an innocent bystander in adaptive evolution; however, there is increasing evidence that mitochondrial DNA (mtDNA) is an important contributor to viability and fecundity. Some of this evidence is now well documented, with mtDNA mutations having been shown to play a causal role in degenerative diseases, ageing, and cancer. However, most research on mtDNA has ignored the possibility that other instances exist where mtDNA mutations could have profound fitness consequences. Recent work in humans and other species now indicates that mtDNA mutations play an important role in sperm function, male fertility, and male fitness. Ironically, deleterious mtDNA mutations that affect only males, such as those that impair sperm function, will not be subject to natural selection because mitochondria are generally maternally inherited and could reach high frequencies in populations if the mutations are not disadvantageous in females. Here, we review how such mtDNA mutations might affect the viability of natural populations. We consider factors that increase or decrease the strength of the effect of mtDNA mutations on population viability and discuss what mechanisms exist to mitigate deleterious mtDNA effects.
Link
May 16, 2006
Weak genealogical link between Etruscans and modern Tuscans
This paragraph from the article echoes some of the sentiments that I have expressed before on this blog and elsewhere:
Serial coalescent simulations suggest a weak genealogical relationship between Etruscans and modern Tuscans
Elise M. S. Belle et al.
The Etruscans, the only preclassical European population that has been genetically characterized so far, share only two haplotypes with their modern geographic counterparts, the Tuscans, who, nonetheless, appear to be their closest relatives. We modeled 10 demographic scenarios spanning the last 2,500 years and tested by serial coalescent simulation whether any are consistent with the patterns of genetic diversity observed within and between the Etruscan and the modern Tuscan populations. Models in which the Etruscans are the direct ancestors of modern Tuscans appear compatible with the observed data only when they also include a very high mutation rate and an ancient founder effect. A better fit was obtained when the ancient and the modern samples were extracted from two independently evolving populations, connected by little migration. Simulated and observed parameters were also similar for a scenario in which the ancient samples came from a subset, e.g., a social elite, genetically differentiated from the bulk of the Etruscan population. In principle, these results may be biased by factors such as gross and systematic errors in the ancient DNA sequences and failure to sample suitable modern individuals. If neither proves to be the case, this study strongly suggests that either the mitochondrial mutation rate is much higher than currently believed or the Etruscans left very few modern mitochondrial descendants.
Link
Regarding time depth, essentially all studies of mtDNA variation in Europe have drawn conclusions regarding demographic phenomena occurring in a rather remote past. Neolithic or even Paleolithic demographic processes have been inferred from patterns in modern mtDNA diversity in the absence of genetic information on past populations (see, e.g., refs. 10 and 27–29) under the implicit assumption of genetic continuity among people dwelling in the same region at different time periods. The results of this study imply that this assumption is not always correct and that theProc. Natl. Acad. Sci. USA, 10.1073/pnas.0509718103
mitochondrial gene pool can undergo a drastic turnover in as few as 100 generations.
Serial coalescent simulations suggest a weak genealogical relationship between Etruscans and modern Tuscans
Elise M. S. Belle et al.
The Etruscans, the only preclassical European population that has been genetically characterized so far, share only two haplotypes with their modern geographic counterparts, the Tuscans, who, nonetheless, appear to be their closest relatives. We modeled 10 demographic scenarios spanning the last 2,500 years and tested by serial coalescent simulation whether any are consistent with the patterns of genetic diversity observed within and between the Etruscan and the modern Tuscan populations. Models in which the Etruscans are the direct ancestors of modern Tuscans appear compatible with the observed data only when they also include a very high mutation rate and an ancient founder effect. A better fit was obtained when the ancient and the modern samples were extracted from two independently evolving populations, connected by little migration. Simulated and observed parameters were also similar for a scenario in which the ancient samples came from a subset, e.g., a social elite, genetically differentiated from the bulk of the Etruscan population. In principle, these results may be biased by factors such as gross and systematic errors in the ancient DNA sequences and failure to sample suitable modern individuals. If neither proves to be the case, this study strongly suggests that either the mitochondrial mutation rate is much higher than currently believed or the Etruscans left very few modern mitochondrial descendants.
Link
May 13, 2006
The origin of European cattle
PNAS has a new article on the origin of European cattle. From the conclusions:
The origin of European cattle: Evidence from modern and ancient DNA
Albano Beja-Pereira et al.
Cattle domestication from wild aurochsen was among the most important innovations during the Neolithic agricultural revolution. The available genetic and archaeological evidence points to at least two major sites of domestication in India and in the Near East, where zebu and the taurine breeds would have emerged independently. Under this hypothesis, all present-day European breeds would be descended from cattle domesticated in the Near East and subsequently spread during the diffusion of herding and farming lifestyles. We present here previously undescribed genetic evidence in contrast with this view, based on mtDNA sequences from five Italian aurochsen dated between 7,000 and 17,000 years B.P. and >1,000 modern cattle from 51 breeds. Our data are compatible with local domestication events in Europe and support at least some levels of introgression from the aurochs in Italy. The distribution of genetic variation in modern cattle suggest also that different south European breeds were affected by introductions from northern Africa. If so, the European cattle may represent a more variable and valuable genetic resource than previously realized, and previous simple hypotheses regarding the domestication process and the diffusion of selected breeds should be revised.
Link
The modern and ancient mtDNA sequences we present here do not support the currently accepted hypothesis of a single Neolithic origin in the Near East. The processes of livestock domestication and diffusion were certainly more complex than previously suggested, and our data provide some evidence in favor of the hypothesis that the origin of European cattle is multiple. Breeds domesticated in the Near East and introduced in Europe during the Neolithic diffusion probably intermixed, at least in some regions, with local wild animals and with African cattle introduced by maritime routes. As a consequence, European breeds should represent a more diverse and important genetic resource than previously recognized, especially in the Southern regions.Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0509210103
The origin of European cattle: Evidence from modern and ancient DNA
Albano Beja-Pereira et al.
Cattle domestication from wild aurochsen was among the most important innovations during the Neolithic agricultural revolution. The available genetic and archaeological evidence points to at least two major sites of domestication in India and in the Near East, where zebu and the taurine breeds would have emerged independently. Under this hypothesis, all present-day European breeds would be descended from cattle domesticated in the Near East and subsequently spread during the diffusion of herding and farming lifestyles. We present here previously undescribed genetic evidence in contrast with this view, based on mtDNA sequences from five Italian aurochsen dated between 7,000 and 17,000 years B.P. and >1,000 modern cattle from 51 breeds. Our data are compatible with local domestication events in Europe and support at least some levels of introgression from the aurochs in Italy. The distribution of genetic variation in modern cattle suggest also that different south European breeds were affected by introductions from northern Africa. If so, the European cattle may represent a more variable and valuable genetic resource than previously realized, and previous simple hypotheses regarding the domestication process and the diffusion of selected breeds should be revised.
Link
May 12, 2006
mtDNA of the last Viking King
Forensic Sci Int. 2006 May 8; [Epub ahead of print]
The last Viking King: A royal maternity case solved by ancient DNA analysis.
Dissing J, Binladen J, Hansen A, Sejrsen B, Willerslev E, Lynnerup N.
The last of the Danish Viking Kings, Sven Estridsen, died in a.d. 1074 and is entombed in Roskilde Cathedral with other Danish kings and queens. Sven's mother, Estrid, is entombed in a pillar across the chancel. However, while there is no reasonable doubt about the identity of Sven, there have been doubts among historians whether the woman entombed was indeed Estrid. To shed light on this problem, we have extracted and analysed mitochondrial DNA (mtDNA) from pulp of teeth from each of the two royals. Four overlapping DNA-fragments covering about 400bp of hypervariable region 1 (HVR-1) of the D-loop were PCR amplified, cloned and a number of clones with each segment were sequenced. Also a segment containing the H/non-H specific nucleotide 7028 was sequenced. Consensus sequences were determined and D-loop results were replicated in an independent laboratory. This allowed the assignment of King Sven Estridsen to haplogroup H; Estrid's sequence differed from that of Sven at two positions in HVR-1, 16093T-->C and 16304T-->C, indicating that she belongs to subgroup H5a. Given the maternal inheritance of mtDNA, offspring will have the same mtDNA sequence as their mother with the exception of rare cases where the sequence has been altered by a germ line mutation. Therefore, the observation of two sequence differences makes it highly unlikely that the entombed woman was the mother of Sven. In addition, physical examination of the skeleton and the teeth strongly indicated that this woman was much younger (approximately 35 years) at the time of death than the 70 years history records tell. Although the entombed woman cannot be the Estrid, she may well be one of Sven's two daughters-in-law who were also called Estrid and who both became queens.
Link
The last Viking King: A royal maternity case solved by ancient DNA analysis.
Dissing J, Binladen J, Hansen A, Sejrsen B, Willerslev E, Lynnerup N.
The last of the Danish Viking Kings, Sven Estridsen, died in a.d. 1074 and is entombed in Roskilde Cathedral with other Danish kings and queens. Sven's mother, Estrid, is entombed in a pillar across the chancel. However, while there is no reasonable doubt about the identity of Sven, there have been doubts among historians whether the woman entombed was indeed Estrid. To shed light on this problem, we have extracted and analysed mitochondrial DNA (mtDNA) from pulp of teeth from each of the two royals. Four overlapping DNA-fragments covering about 400bp of hypervariable region 1 (HVR-1) of the D-loop were PCR amplified, cloned and a number of clones with each segment were sequenced. Also a segment containing the H/non-H specific nucleotide 7028 was sequenced. Consensus sequences were determined and D-loop results were replicated in an independent laboratory. This allowed the assignment of King Sven Estridsen to haplogroup H; Estrid's sequence differed from that of Sven at two positions in HVR-1, 16093T-->C and 16304T-->C, indicating that she belongs to subgroup H5a. Given the maternal inheritance of mtDNA, offspring will have the same mtDNA sequence as their mother with the exception of rare cases where the sequence has been altered by a germ line mutation. Therefore, the observation of two sequence differences makes it highly unlikely that the entombed woman was the mother of Sven. In addition, physical examination of the skeleton and the teeth strongly indicated that this woman was much younger (approximately 35 years) at the time of death than the 70 years history records tell. Although the entombed woman cannot be the Estrid, she may well be one of Sven's two daughters-in-law who were also called Estrid and who both became queens.
Link
"sex" on Google Trends
Foreign Dispatches links to the Google Trends query on "sex", finding that users of the following languages query on "sex" most often:
Similarly, young people are perhaps more interested in sex than older ones. With similar reasoning, we can see that in countries where old people use the Internet, the apparent interest in sex will be lower than in ones where Internet usage is limited to young people.
Furthermore, the cost of accessing the Internet is not similar across countries. In the First World, nearly everyone can be online, but in poorer countries Internet access is limited to the more affluent sections of society, i.e., working-age males.
And of course, "sex" is an English word, so someone who queries on "sex" must have at least a basic knowledge of English. Once again, the group of people who know some English is not the same as that which does not, biasing the perceived interest in the subject. Not to mention that one's level of English knowledge will dictate their use of the query term "sex" or not: poor English speakers may search for "sex", while more knowledgeable speakers may use one of the more specific terms for what they are searching for.
To summarize, the intensity of querying on a topic (and "sex" is just a convenient example) is a function not only of interest in the topic, but also of the characteristics of the querying population.
The author of Foreign Dispatches explains this as follows:
- Arabic
- Vietnamese
- Turkish
- Polish
- Romanian
For Arabic, Vietnamese, Turkish, Polish and Romanian to figure in the top 5 without their users even numbering amongst the top 10 best represented groups online says very clearly that the intensity of the online interest in the topic of sex is several orders of magnitude larger than elsewhere.This is a good example of faulty reasoning. To see why this is the case, imagine that M out of 100 queries submitted by males are on the topic of "sex", while F out of 100 queries submitted by females are on the same topic. If males are more likely to search on this topic than females, then countries that are more male-dominated (i.e., in which women don't use computers as much) will appear to be more "sex"-interested. For example, if M=5 and F=1, then a country where 60% of Internet users are male, will have an observed interest of 3.4, whereas if 90% of Internet users are male, then the corresponding interest will be 4.6.
Similarly, young people are perhaps more interested in sex than older ones. With similar reasoning, we can see that in countries where old people use the Internet, the apparent interest in sex will be lower than in ones where Internet usage is limited to young people.
Furthermore, the cost of accessing the Internet is not similar across countries. In the First World, nearly everyone can be online, but in poorer countries Internet access is limited to the more affluent sections of society, i.e., working-age males.
And of course, "sex" is an English word, so someone who queries on "sex" must have at least a basic knowledge of English. Once again, the group of people who know some English is not the same as that which does not, biasing the perceived interest in the subject. Not to mention that one's level of English knowledge will dictate their use of the query term "sex" or not: poor English speakers may search for "sex", while more knowledgeable speakers may use one of the more specific terms for what they are searching for.
To summarize, the intensity of querying on a topic (and "sex" is just a convenient example) is a function not only of interest in the topic, but also of the characteristics of the querying population.
May 11, 2006
Skin color sexual dimorphism in humans
Skin color sexual dimorphism in humans involves the lighter pigmentation of females compared to males. An explanation for this phenomenon is that males tend to favor lighter-skinned females. A prediction of this theory is that sexual dimorphism should be more pronounced in regions of the world with low solar radiation. Skin color is under selection close to the equator, with such populations being constrained to be dark in order to shield themselves from bright sunlight. Conversely, in areas of low solar radiation, this selective constraint should be relaxed, and as a result, the postulated preference for light skin in women would become more important. This would manifest itself as greater sexual dimorphism in such regions.
The authors of this paper tested this prediction directly, by comparing skin reflectance date of adult men and women from different populations with latitude, discovering that sexual dimorphism is not correlated with latitude. Hence, one of the predictions of the "sexual selection hypothesis" is not supported by the data.
From the conclusions of the paper:
American Journal of Physical Anthropology (Early View)
Human skin-color sexual dimorphism: A test of the sexual selection hypothesis
Lorena Madrigal, William Kelly
Abstract
Applied to skin color, the sexual selection hypothesis proposes that male preference for light-skinned females explains the presence of light skin in areas of low solar radiation. According to this proposal, in areas of high solar radiation, natural selection for dark skin overrides the universal preference of males for light females. But in areas in which natural selection ceases to act, sexual selection becomes more important, and causes human populations to become light-skinned, and females to be lighter than males. The sexual selection hypothesis proposes that human sexual dimorphism of skin color should be positively correlated with distance from the equator. We tested the prediction that sexual dimorphism should increase with increasing latitude, using adult-only data sets derived from measurements with standard reflectance spectrophotometric devices. Our analysis failed to support the prediction of a positive correlation between increasing distance from the equator and increased sexual dimorphism. We found no evidence in support of the sexual selection hypothesis.
Link
The authors of this paper tested this prediction directly, by comparing skin reflectance date of adult men and women from different populations with latitude, discovering that sexual dimorphism is not correlated with latitude. Hence, one of the predictions of the "sexual selection hypothesis" is not supported by the data.
From the conclusions of the paper:
We tested the hypothesis that human sexual dimorphism
of skin color should be positively correlated with
distance from the equator, a proposal generated by the
sexual selection hypothesis. We found no support for that
proposition. Before this paper was written, the sexual
selection hypothesis was based on stated male-preference
data in a number of human groups. Here, we focused on
the actual pattern of sexual dimorphism. We report that
the distribution of human sexual dimorphism in relation
to latitude is not that which is predicted by the sexual
selection hypothesis. According to that hypothesis, in
areas of low solar radiation, there should be greater sexual
dimorphism, because sexual selection for lighter
females is not counterbalanced by natural selection
for dark skin. Our data analysis does not support this prediction.
American Journal of Physical Anthropology (Early View)
Human skin-color sexual dimorphism: A test of the sexual selection hypothesis
Lorena Madrigal, William Kelly
Abstract
Applied to skin color, the sexual selection hypothesis proposes that male preference for light-skinned females explains the presence of light skin in areas of low solar radiation. According to this proposal, in areas of high solar radiation, natural selection for dark skin overrides the universal preference of males for light females. But in areas in which natural selection ceases to act, sexual selection becomes more important, and causes human populations to become light-skinned, and females to be lighter than males. The sexual selection hypothesis proposes that human sexual dimorphism of skin color should be positively correlated with distance from the equator. We tested the prediction that sexual dimorphism should increase with increasing latitude, using adult-only data sets derived from measurements with standard reflectance spectrophotometric devices. Our analysis failed to support the prediction of a positive correlation between increasing distance from the equator and increased sexual dimorphism. We found no evidence in support of the sexual selection hypothesis.
Link
mtDNA from medieval al-Andalus
Am J Phys Anthropol. 2006 May 9; [Epub ahead of print]
Human mitochondrial DNA diversity in an archaeological site in al-Andalus: Genetic impact of migrations from North Africa in medieval Spain.
Casas MJ, Hagelberg E, Fregel R, Larruga JM, Gonzalez AM.
Mitochondrial DNA sequences and restriction fragment polymorphisms were retrieved from three Islamic 12th-13th century samples of 71 bones and teeth (with >85% efficiency) from Madinat Baguh (today called Priego de Cordoba, Spain). Compared with 108 saliva samples from the present population of the same area, the medieval samples show a higher proportion of sub-Saharan African lineages that can only partially be attributed to the historic Muslim occupation. In fact, the unique sharing of transition 16175, in L1b lineages, with Europeans, instead of Africans, suggests a more ancient arrival to Europe from Africa. The present day Priego sample is more similar to the current south Iberian population than to the medieval sample from the same area. The increased gene flow in modern times could be the main cause of this difference.
Link
Human mitochondrial DNA diversity in an archaeological site in al-Andalus: Genetic impact of migrations from North Africa in medieval Spain.
Casas MJ, Hagelberg E, Fregel R, Larruga JM, Gonzalez AM.
Mitochondrial DNA sequences and restriction fragment polymorphisms were retrieved from three Islamic 12th-13th century samples of 71 bones and teeth (with >85% efficiency) from Madinat Baguh (today called Priego de Cordoba, Spain). Compared with 108 saliva samples from the present population of the same area, the medieval samples show a higher proportion of sub-Saharan African lineages that can only partially be attributed to the historic Muslim occupation. In fact, the unique sharing of transition 16175, in L1b lineages, with Europeans, instead of Africans, suggests a more ancient arrival to Europe from Africa. The present day Priego sample is more similar to the current south Iberian population than to the medieval sample from the same area. The increased gene flow in modern times could be the main cause of this difference.
Link
Y-haplogroups and prostate cancer in Japan
Prostate Cancer Prostatic Dis. 2006 May 9;
Prostate cancer incidence varies among males from different Y-chromosome lineages.
Ewis AA, Lee J, Naroda T, Sano T, Kagawa S, Iwamoto T, Shinka T, Shinohara Y, Ishikawa M, Baba Y, Nakahori Y
The incidence rate of prostate cancer in African-American males is two times higher than Caucasian men and ten times higher than Japanese men. The geographical specificity of Y haplogroups implies that males from different ethnic groups undoubtedly have various Y lineages with different Y-chromosomal characteristics that may affect their susceptibility or resistance to such a male-specific cancer. To confirm this hypothesis we studied the Y-chromosomal haplogroups of 92 Japanese prostate cancer patients comparing them with randomly selected 109 unrelated healthy Japanese male controls who were confirmed to be residents of the same geographical area. Males could be classified using three binary Y-chromosome markers (sex-determining region Y (SRY), YAP, 47z) into four haplogroups DE, O2b(*), O2b1, and untagged group. Our results confirmed that prostate cancer incidence varies among males from different Y-chromosome lineages. Males from DE and the untagged haplogroups are at a significantly higher risk to develop prostate cancer than O2b(*) and O2b1 haplogroups (P=0.01), odds ratio 2.17 and 95% confidence interval (1.16-4.07). Males from haplogroup DE are over-represented in the patient group showing a percentage of 41.3%. The underlying possible causes of susceptibility variations of different Y lineages for such a male-specific cancer tumorigenesis are discussed. These findings explain the lower incidence of prostate cancer in Japanese and other South East Asian males than other populations. To our knowledge, this is the first reliable study examining the association between prostate cancer and Y-chromosomal haplogroups, comparing prostate cancer patients with carefully selected matched controls.
Link
Prostate cancer incidence varies among males from different Y-chromosome lineages.
Ewis AA, Lee J, Naroda T, Sano T, Kagawa S, Iwamoto T, Shinka T, Shinohara Y, Ishikawa M, Baba Y, Nakahori Y
The incidence rate of prostate cancer in African-American males is two times higher than Caucasian men and ten times higher than Japanese men. The geographical specificity of Y haplogroups implies that males from different ethnic groups undoubtedly have various Y lineages with different Y-chromosomal characteristics that may affect their susceptibility or resistance to such a male-specific cancer. To confirm this hypothesis we studied the Y-chromosomal haplogroups of 92 Japanese prostate cancer patients comparing them with randomly selected 109 unrelated healthy Japanese male controls who were confirmed to be residents of the same geographical area. Males could be classified using three binary Y-chromosome markers (sex-determining region Y (SRY), YAP, 47z) into four haplogroups DE, O2b(*), O2b1, and untagged group. Our results confirmed that prostate cancer incidence varies among males from different Y-chromosome lineages. Males from DE and the untagged haplogroups are at a significantly higher risk to develop prostate cancer than O2b(*) and O2b1 haplogroups (P=0.01), odds ratio 2.17 and 95% confidence interval (1.16-4.07). Males from haplogroup DE are over-represented in the patient group showing a percentage of 41.3%. The underlying possible causes of susceptibility variations of different Y lineages for such a male-specific cancer tumorigenesis are discussed. These findings explain the lower incidence of prostate cancer in Japanese and other South East Asian males than other populations. To our knowledge, this is the first reliable study examining the association between prostate cancer and Y-chromosomal haplogroups, comparing prostate cancer patients with carefully selected matched controls.
Link
Worldwide Neolithic Demographic Transition
Current Anthropology, volume 47 (2006), pages 341–365
Testing the Hypothesis of a Worldwide Neolithic Demographic Transition
Jean-Pierre Bocquet-Appel and Stephan Naji
The signal of a major demographic change characterized by a relatively abrupt increase in the proportion of immature skeletons has been detected in a paleoanthropological database of 38 Mesolithic-Neolithic cemeteries from Europe and North Africa. From the Mesolithic to the Neolithic, the proportion of immature skeletons increases by 20 30% over a period of 500700 years, indicating a notable increase in the crude birth rate. This shift has been called the Neolithic demographic transition. A similar signal has been detected in an independent set of archaeological data, namely, enclosures. This paper presents results from a sample of 62 cemeteries in North America (7,755 BP350 BP) that point to the same transition over a period of 600800 years.
30% over a period of 500700 years, indicating a notable increase in the crude birth rate. This shift has been called the Neolithic demographic transition. A similar signal has been detected in an independent set of archaeological data, namely, enclosures. This paper presents results from a sample of 62 cemeteries in North America (7,755 BP350 BP) that point to the same transition over a period of 600800 years.
Link
Testing the Hypothesis of a Worldwide Neolithic Demographic Transition
Jean-Pierre Bocquet-Appel and Stephan Naji
The signal of a major demographic change characterized by a relatively abrupt increase in the proportion of immature skeletons has been detected in a paleoanthropological database of 38 Mesolithic-Neolithic cemeteries from Europe and North Africa. From the Mesolithic to the Neolithic, the proportion of immature skeletons increases by 20
30% over a period of 500700 years, indicating a notable increase in the crude birth rate. This shift has been called the Neolithic demographic transition. A similar signal has been detected in an independent set of archaeological data, namely, enclosures. This paper presents results from a sample of 62 cemeteries in North America (7,755 BP350 BP) that point to the same transition over a period of 600800 years.Link
May 10, 2006
Anthropological types of Corded Ware and Yamna cultures
Some useful descriptions from the paper (pp. 351-353):
See also a previous related post on the Anthropology of Sredny Stog and Novodanylovka.
Journal of Indo-European Studies Vol. 33: 3-4, p. 339
The Yamna Culture and the Indo-European Homeland Problem
D. Ya. Telegin
Excavations between the rivers Orel' and Samara have uncovered burials of a syncretic nature that attest contacts between the spheres of the Corded Ware and Yamna cultures. It is suggested that these may indicate early contacts between proto-Indo-Iranians and the prehistoric ancestors of the Balts and Slavs.
The Corded Ware culture includes about twenty variants (Sveshnikov 1974) and occupies predominantly Central Europe, Scandinavia, Sub-Carpathia and also the Upper Volga (Fig. 1). It was formed in Central Europe, in an area of special concentration of qute distinct cultures of different origins, including those from Mediterranean and North-Baltic regions of Europe. Bearers of the southern LBK (Roessen, Tisza, Lengyel, etc) were engaged in cattle-breeding, had developed ceramic production, and were familiar with plastic arts. They buried their dead in the flexed position on the right or left side. Generally it was a comparatively gracile short people of the Mediterranean (South Europoid) anthropological type.
Peoples of the Baltic circle of cultures (Ertebo/lle) were hunters and fishermen, and produced only one or two pottery forms. This was a rather tall, broad-faced population of the North Europoid type, who buried their dead in the extended position on the back.
Certain cultures of syncretic appearance involving Northern and Southern features were formed in Central Europe and the Baltic during the 4th-3rd millennium BC, e.g. Comb Ware, TRB, and Globular Amphora cultures. During the Early Bronze Age these cultures were displaced by the Corded Ware (Battle Axe) culture characterized by flexed inhumation on the back or side under a barrow. Specifically, this culture embraces both bottle-like vessels and bowls with funnel-like neck of the Northern circle of cultures, and also vessels of the Danubian type. In an anthropological sense this population combines traits of southern gracile and northern massive types, in particular bearers of TRB culture (Schwidetzky 1978).
...
The Yamna culture of the Pontic-Caspian steppe is recorded for an enormous territory between the North-Western Pontic area and Trans-Uralia. Its sites are known here in the basin of the Emba and Tobol rivers, the Karaganda region and further eastward (Merpert 1974). The Yamna population generally belongs to the European race. It was tall (175.5cm), dolichocephalic, with broad faces of medium height. Among them there were, however, more robust elements with high and wide faces of the proto-Europoid type, and also more gracile individuals with narrow and high faces, probably reflecting contacts with the East Mediterranean type (Kurts 1984: 90).
See also a previous related post on the Anthropology of Sredny Stog and Novodanylovka.
Journal of Indo-European Studies Vol. 33: 3-4, p. 339
The Yamna Culture and the Indo-European Homeland Problem
D. Ya. Telegin
Excavations between the rivers Orel' and Samara have uncovered burials of a syncretic nature that attest contacts between the spheres of the Corded Ware and Yamna cultures. It is suggested that these may indicate early contacts between proto-Indo-Iranians and the prehistoric ancestors of the Balts and Slavs.
May 08, 2006
Neanderthals and Humans
LiveScience has an interesting article referring to Paul Mellars' latest research, which I covered a few months ago:
See also the interesting timeline of human evolution in the article.
The overlap figure shrank in February with new research by Paul Mellars of Cambridge University based on improved carbon-14 dating to show that modern humans started encroaching from Israel upon Neanderthal territory in the Balkans 3,000 years sooner than previously thought. This rate suggests Neanderthals succumbed sooner to big climate shifts or competition from modern humans for resources and that they might have overlapped for only 1,000 years at sites in western France.
Try zero years, says anthropologist John Hawks of the University of Wisconsin-Madison.
There is no longer any biological evidence of overlap between Neanderthals and non-Neanderthals in Europe, Hawks wrote recently in his blog. Many anthropologists are aware of this but "would like to sweep it under a rug," Hawks told LiveScience.
See also the interesting timeline of human evolution in the article.
May 06, 2006
The Slavs in Russia
I believe that this is related to my recent post on Y chromosomes and mtDNA of Russians.
How the Slavs conquered Russia (Informnauka (Informscience) Agency):
Geneticist specialists from the Institute of Biological Problems of the North, Far-East Branch of Russian Academy of Sciences, are reconstructing the picture of Eurasia colonization by the Slavs. According to the researchers’ opinion, the Slavonic men and women jointly developed the territory of the south of contemporary Russia. However, after the 9th century, women used to stay at home, and colonization of the east and north was mainly performed by men.
This conclusion was made by geneticists through analyzing variable consecutions of DNA of mitochondria and of some sections of Y-chromosome with representatives of 10 Russian populations from the Stavropol Territory in the south through the Pskov Region in the north and from the Orel Region in the west through the Nizhni Novgorod Region in the east. The mitochondrial DNA is inherited from generation to generation along a female line, DNA of Y- chromosome – along a male line. Analysis of variability of these consecutions allows to judge about migrations of our forefathers and foremothers.
Along the maternal line, Russian populations are rather close to each other. They can be conditionally split into two zones. Inhabitants of the south-eastern zone (including the Orel, Rostov, Kursk, Kaluga and Saratov Regions and the Stavropol Territory) have the roots among western Slavs, Baltic and some Finno-Ugric nations (Poles, Lithuanians and Estonians). Ancestors of Russians in the north-eastern zone took wives from the Finno-Ugric and other nations of Eastern Europe (Finns, Karelians, Maris, Tatars and Adygeis).
Comparison of Y-chromosomes of Russian populations provides different results. Only the Pskov and coast-dweller populations are close along the paternal line to the Finno-Ugric and Baltic nations of the Northern and Eastern Europe, the overwhelming majority of Russians are relatives to the Poles, Ukrainians and Byelorussians. Judging by consecution variations of the “male” chromosome, the Slavs, arriving in various locations of Eastern Europe, contacted in different ways with residential population: in some places - closely and in some others – otherwise.
Genetic analysis results agree with the anthropological data, according to which Russian populations can be divided into three zones. In the western part of the ethnic territory, Russians descend from the Slavs who had come from Central Europe. Russian population of the central part appeared as a result of mixture of the Slavs with the Finno-Ugric nations, Eastern European mothers dominating in these populations, and the population of the North evidently has in its genealogy Finno-Ugric ancestors of both sexes. According to the geneticists, the reasons for these differences are caused by different participation of men and women in Slavonic migrations. Women apparently participated only in early phases of the Slavs’ migration into Eastern Europe. Starting from the 9th century, colonization of the east and the north of Eastern Europe was mainly performed by men who chose wives from residential population. The obtained picture needs more precise definition, therefore the researchers are planning to further investigate variability of specific “maternal” and “ paternal” DNAs in different Russian populations.
How the Slavs conquered Russia (Informnauka (Informscience) Agency):
Geneticist specialists from the Institute of Biological Problems of the North, Far-East Branch of Russian Academy of Sciences, are reconstructing the picture of Eurasia colonization by the Slavs. According to the researchers’ opinion, the Slavonic men and women jointly developed the territory of the south of contemporary Russia. However, after the 9th century, women used to stay at home, and colonization of the east and north was mainly performed by men.
This conclusion was made by geneticists through analyzing variable consecutions of DNA of mitochondria and of some sections of Y-chromosome with representatives of 10 Russian populations from the Stavropol Territory in the south through the Pskov Region in the north and from the Orel Region in the west through the Nizhni Novgorod Region in the east. The mitochondrial DNA is inherited from generation to generation along a female line, DNA of Y- chromosome – along a male line. Analysis of variability of these consecutions allows to judge about migrations of our forefathers and foremothers.
Along the maternal line, Russian populations are rather close to each other. They can be conditionally split into two zones. Inhabitants of the south-eastern zone (including the Orel, Rostov, Kursk, Kaluga and Saratov Regions and the Stavropol Territory) have the roots among western Slavs, Baltic and some Finno-Ugric nations (Poles, Lithuanians and Estonians). Ancestors of Russians in the north-eastern zone took wives from the Finno-Ugric and other nations of Eastern Europe (Finns, Karelians, Maris, Tatars and Adygeis).
Comparison of Y-chromosomes of Russian populations provides different results. Only the Pskov and coast-dweller populations are close along the paternal line to the Finno-Ugric and Baltic nations of the Northern and Eastern Europe, the overwhelming majority of Russians are relatives to the Poles, Ukrainians and Byelorussians. Judging by consecution variations of the “male” chromosome, the Slavs, arriving in various locations of Eastern Europe, contacted in different ways with residential population: in some places - closely and in some others – otherwise.
Genetic analysis results agree with the anthropological data, according to which Russian populations can be divided into three zones. In the western part of the ethnic territory, Russians descend from the Slavs who had come from Central Europe. Russian population of the central part appeared as a result of mixture of the Slavs with the Finno-Ugric nations, Eastern European mothers dominating in these populations, and the population of the North evidently has in its genealogy Finno-Ugric ancestors of both sexes. According to the geneticists, the reasons for these differences are caused by different participation of men and women in Slavonic migrations. Women apparently participated only in early phases of the Slavs’ migration into Eastern Europe. Starting from the 9th century, colonization of the east and the north of Eastern Europe was mainly performed by men who chose wives from residential population. The obtained picture needs more precise definition, therefore the researchers are planning to further investigate variability of specific “maternal” and “ paternal” DNAs in different Russian populations.
Racial admixture, body mass index, and blood pressure
It is nice to see this article referring to "racial admixture" rather than one of the many euphemisms used to describe descent of individuals from two or more differentiated ancestral groups.
Human Genetics (Online first)
Racial admixture and its impact on BMI and blood pressure in African and Mexican Americans
Hua Tang et al.
Abstract Admixed populations such as African Americans and Hispanic Americans present both challenges and opportunities in genetic epidemiologic research. Because of variation in admixture levels among individuals, case-control association studies may be subject to stratification bias. On the other hand, admixed populations also present special opportunities both for examining the role of genetic and environmental factors for observed racial/ethnic differences, and for possibly mapping alleles that contribute to such differences. Here we examined the distribution and relationship of individual admixture (IA) estimates with BMI and three measures of blood pressure in two admixed populations in the NHLBI Family Blood Pressure Program (FBPP): African Americans and Mexican Americans. For the African Americans, we observed modest but significant differences in average African IA among four recruitment sites. We observed a slight excess of African IA among hypertensives compared to normotensives, and a positive (non-significant) regression of African IA on blood pressure in untreated participants. Within Mexican Americans, we found no difference in average IA between hypertensives and normotensives, but a positive (marginally significant) regression of African IA on diastolic blood pressure. We also observed a significant positive regression of Caucasian IA (and negative regression of Native American IA) on BMI. Our results are suggestive of genetic differences between Africans and non-Africans that influence blood pressure, but such effects are likely to be modest compared to environmental ones. Excess obesity among Native Americans compared to whites is not consistent with a simple genetic explanation.
Link
Human Genetics (Online first)
Racial admixture and its impact on BMI and blood pressure in African and Mexican Americans
Hua Tang et al.
Abstract Admixed populations such as African Americans and Hispanic Americans present both challenges and opportunities in genetic epidemiologic research. Because of variation in admixture levels among individuals, case-control association studies may be subject to stratification bias. On the other hand, admixed populations also present special opportunities both for examining the role of genetic and environmental factors for observed racial/ethnic differences, and for possibly mapping alleles that contribute to such differences. Here we examined the distribution and relationship of individual admixture (IA) estimates with BMI and three measures of blood pressure in two admixed populations in the NHLBI Family Blood Pressure Program (FBPP): African Americans and Mexican Americans. For the African Americans, we observed modest but significant differences in average African IA among four recruitment sites. We observed a slight excess of African IA among hypertensives compared to normotensives, and a positive (non-significant) regression of African IA on blood pressure in untreated participants. Within Mexican Americans, we found no difference in average IA between hypertensives and normotensives, but a positive (marginally significant) regression of African IA on diastolic blood pressure. We also observed a significant positive regression of Caucasian IA (and negative regression of Native American IA) on BMI. Our results are suggestive of genetic differences between Africans and non-Africans that influence blood pressure, but such effects are likely to be modest compared to environmental ones. Excess obesity among Native Americans compared to whites is not consistent with a simple genetic explanation.
Link
May 04, 2006
Faster evolution in tropical climates
See also Evolution Occurs Faster at the Equator at LiveScience.
PNAS (early view)
The road from Santa Rosalia: A faster tempo of evolution in tropical climates
Shane Wright et al.
Using an appropriately designed and replicated study of a latitudinal influence on rates of evolution, we test the prediction by K. Rohde [(1992) Oikos 65, 514-527] that the tempo of molecular evolution in the tropics is greater than at higher latitudes. Consistent with this prediction we found tropical plant species had more than twice the rate of molecular evolution as closely related temperate congeners. Rohde's climate-speciation hypothesis constitutes one explanation for the cause of that relationship. This hypothesis suggests that mutagenesis occurs more frequently as productivity and metabolic rates increase toward the equator. More rapid mutagenesis was then proposed as the mechanism that increases evolutionary tempo and rates of speciation. A second possible explanation is that faster rates of molecular evolution result from higher tropical speciation rates [e.g., Bromham, L. & Cardillo, M. (2003) J. Evol. Biol. 16, 200-207]. However, we found the relationship continued to hold for genera with the same number of, or more, species in temperate latitudes. This finding suggests that greater rates of speciation in the tropics do not cause higher rates of molecular evolution. A third explanation is that more rapid genetic drift might have occurred in smaller tropical species populations [Stevens, G. C. (1989) Am. Nat. 133, 240-256]. However, we targeted common species to limit the influence of genetic drift, and many of the tropical species we used, despite occurring in abundant populations, had much higher rates of molecular evolution. Nonetheless, this issue is not completely resolved by that precaution and requires further examination.
Link
PNAS (early view)
The road from Santa Rosalia: A faster tempo of evolution in tropical climates
Shane Wright et al.
Using an appropriately designed and replicated study of a latitudinal influence on rates of evolution, we test the prediction by K. Rohde [(1992) Oikos 65, 514-527] that the tempo of molecular evolution in the tropics is greater than at higher latitudes. Consistent with this prediction we found tropical plant species had more than twice the rate of molecular evolution as closely related temperate congeners. Rohde's climate-speciation hypothesis constitutes one explanation for the cause of that relationship. This hypothesis suggests that mutagenesis occurs more frequently as productivity and metabolic rates increase toward the equator. More rapid mutagenesis was then proposed as the mechanism that increases evolutionary tempo and rates of speciation. A second possible explanation is that faster rates of molecular evolution result from higher tropical speciation rates [e.g., Bromham, L. & Cardillo, M. (2003) J. Evol. Biol. 16, 200-207]. However, we found the relationship continued to hold for genera with the same number of, or more, species in temperate latitudes. This finding suggests that greater rates of speciation in the tropics do not cause higher rates of molecular evolution. A third explanation is that more rapid genetic drift might have occurred in smaller tropical species populations [Stevens, G. C. (1989) Am. Nat. 133, 240-256]. However, we targeted common species to limit the influence of genetic drift, and many of the tropical species we used, despite occurring in abundant populations, had much higher rates of molecular evolution. Nonetheless, this issue is not completely resolved by that precaution and requires further examination.
Link
Subscribe to:
Posts (Atom)