PLoS ONE doi:10.1371/journal.pone.0005753
The Multifaceted Origin of Taurine Cattle Reflected by the Mitochondrial Genome
Alessandro Achilli et al.
Abstract
A Neolithic domestication of taurine cattle in the Fertile Crescent from local aurochsen (Bos primigenius) is generally accepted, but a genetic contribution from European aurochsen has been proposed. Here we performed a survey of a large number of taurine cattle mitochondrial DNA (mtDNA) control regions from numerous European breeds confirming the overall clustering within haplogroups (T1, T2 and T3) of Near Eastern ancestry, but also identifying eight mtDNAs (1.3%) that did not fit in haplogroup T. Sequencing of the entire mitochondrial genome showed that four mtDNAs formed a novel branch (haplogroup R) which, after the deep bifurcation that gave rise to the taurine and zebuine lineages, constitutes the earliest known split in the mtDNA phylogeny of B. primigenius. The remaining four mtDNAs were members of the recently discovered haplogroup Q. Phylogeographic data indicate that R mtDNAs were derived from female European aurochsen, possibly in the Italian Peninsula, and sporadically included in domestic herds. In contrast, the available data suggest that Q mtDNAs and T subclades were involved in the same Neolithic event of domestication in the Near East. Thus, the existence of novel (and rare) taurine haplogroups highlights a multifaceted genetic legacy from distinct B. primigenius populations. Taking into account that the maternally transmitted mtDNA tends to underestimate the extent of gene flow from European aurochsen, the detection of the R mtDNAs in autochthonous breeds, some of which are endangered, identifies an unexpected reservoir of genetic variation that should be carefully preserved.
Link

May 29, 2009
Ancient mtDNA and craniometric evolution of Amerindians
This paper shows that while the mtDNA gene pool of Amerindians did not undergo substantial change since the Holocene (haplogroups B, C, D were detected in the ancient samples, all of them common today), their cranial morphology changed from a more generalized to a more Mongoloid pattern.
In my opinion, the fact that Amerindians evolved in a Mongoloid direction may suggest one of three things:
Why the type became so successful remains to be seen; adaptive explanations for a rounder skull, flatter face, and heavy eyelids have been proposed as responses to extreme arctic cold, but why would similar phenotypes be selected for in regions of less extreme climate?
Sexual selection might play a role, although it would be difficult to establish over such a large area.
My guess is that various aspects of the Mongoloid pattern existed in low frequency or as isolated tendencies across East Eurasia and America. As populations grew during the Holocene, these traits spread in a wider range. Naturally, in the periphery, their blending was incomplete, with different quasi-Mongoloid types emerging there, e.g., prominent-nosed, round-headed Amerindians vs. flat-nosed, long-headed Proto-Uralics.
Thus, ancestral Amerindians either already had, or later received -by limited gene flow- a set of Mongoloid traits from Asia, which were selected for the same reasons as they did in Asia, but the "bottleneck" of the Bering did not allow them to receive the full package of traits.
PLoS ONE doi:10.1371/journal.pone.0005746
Discrepancy between Cranial and DNA Data of Early Americans: Implications for American Peopling
S. Ivan Perez et al.
Abstract
Currently, one of the major debates about the American peopling focuses on the number of populations that originated the biological diversity found in the continent during the Holocene. The studies of craniometric variation in American human remains dating from that period have shown morphological differences between the earliest settlers of the continent and some of the later Amerindian populations. This led some investigators to suggest that these groups—known as Paleomericans and Amerindians respectively—may have arisen from two biologically different populations. On the other hand, most DNA studies performed over extant and ancient populations suggest a single migration of a population from Northeast Asia. Comparing craniometric and mtDNA data of diachronic samples from East Central Argentina dated from 8,000 to 400 years BP, we show here that even when the oldest individuals display traits attributable to Paleoamerican crania, they present the same mtDNA haplogroups as later populations with Amerindian morphology. A possible explanation for these results could be that the craniofacial differentiation was a local phenomenon resulting from random (i.e. genetic drift) and non-random factors (e.g. selection and plasticity). Local processes of morphological differentiation in America are a probable scenario if we take into consideration the rapid peopling and the great ecological diversity of this continent; nevertheless we will discuss alternative explanations as well.
Link
In my opinion, the fact that Amerindians evolved in a Mongoloid direction may suggest one of three things:
- proto-Mongoloid traits were present as tendencies in the founding population, and they evolved in parallel in the Americas and in East Asia
- proto-Mongoloid traits were absent in the founding population, and they evolved independently in the Americas
- proto-Mongoloid traits were absent in the founding population, but they were added by limited gene flow from Asia
Why the type became so successful remains to be seen; adaptive explanations for a rounder skull, flatter face, and heavy eyelids have been proposed as responses to extreme arctic cold, but why would similar phenotypes be selected for in regions of less extreme climate?
Sexual selection might play a role, although it would be difficult to establish over such a large area.
My guess is that various aspects of the Mongoloid pattern existed in low frequency or as isolated tendencies across East Eurasia and America. As populations grew during the Holocene, these traits spread in a wider range. Naturally, in the periphery, their blending was incomplete, with different quasi-Mongoloid types emerging there, e.g., prominent-nosed, round-headed Amerindians vs. flat-nosed, long-headed Proto-Uralics.
Thus, ancestral Amerindians either already had, or later received -by limited gene flow- a set of Mongoloid traits from Asia, which were selected for the same reasons as they did in Asia, but the "bottleneck" of the Bering did not allow them to receive the full package of traits.
PLoS ONE doi:10.1371/journal.pone.0005746
Discrepancy between Cranial and DNA Data of Early Americans: Implications for American Peopling
S. Ivan Perez et al.
Abstract
Currently, one of the major debates about the American peopling focuses on the number of populations that originated the biological diversity found in the continent during the Holocene. The studies of craniometric variation in American human remains dating from that period have shown morphological differences between the earliest settlers of the continent and some of the later Amerindian populations. This led some investigators to suggest that these groups—known as Paleomericans and Amerindians respectively—may have arisen from two biologically different populations. On the other hand, most DNA studies performed over extant and ancient populations suggest a single migration of a population from Northeast Asia. Comparing craniometric and mtDNA data of diachronic samples from East Central Argentina dated from 8,000 to 400 years BP, we show here that even when the oldest individuals display traits attributable to Paleoamerican crania, they present the same mtDNA haplogroups as later populations with Amerindian morphology. A possible explanation for these results could be that the craniofacial differentiation was a local phenomenon resulting from random (i.e. genetic drift) and non-random factors (e.g. selection and plasticity). Local processes of morphological differentiation in America are a probable scenario if we take into consideration the rapid peopling and the great ecological diversity of this continent; nevertheless we will discuss alternative explanations as well.
Link
Brain structure and IQ
PLoS Comput Biol doi:10.1371/journal.pcbi.1000395
Brain Anatomical Network and Intelligence
Yonghui Li et al.
Abstract
Intuitively, higher intelligence might be assumed to correspond to more efficient information transfer in the brain, but no direct evidence has been reported from the perspective of brain networks. In this study, we performed extensive analyses to test the hypothesis that individual differences in intelligence are associated with brain structural organization, and in particular that higher scores on intelligence tests are related to greater global efficiency of the brain anatomical network. We constructed binary and weighted brain anatomical networks in each of 79 healthy young adults utilizing diffusion tensor tractography and calculated topological properties of the networks using a graph theoretical method. Based on their IQ test scores, all subjects were divided into general and high intelligence groups and significantly higher global efficiencies were found in the networks of the latter group. Moreover, we showed significant correlations between IQ scores and network properties across all subjects while controlling for age and gender. Specifically, higher intelligence scores corresponded to a shorter characteristic path length and a higher global efficiency of the networks, indicating a more efficient parallel information transfer in the brain. The results were consistently observed not only in the binary but also in the weighted networks, which together provide convergent evidence for our hypothesis. Our findings suggest that the efficiency of brain structural organization may be an important biological basis for intelligence.
Link
Brain Anatomical Network and Intelligence
Yonghui Li et al.
Abstract
Intuitively, higher intelligence might be assumed to correspond to more efficient information transfer in the brain, but no direct evidence has been reported from the perspective of brain networks. In this study, we performed extensive analyses to test the hypothesis that individual differences in intelligence are associated with brain structural organization, and in particular that higher scores on intelligence tests are related to greater global efficiency of the brain anatomical network. We constructed binary and weighted brain anatomical networks in each of 79 healthy young adults utilizing diffusion tensor tractography and calculated topological properties of the networks using a graph theoretical method. Based on their IQ test scores, all subjects were divided into general and high intelligence groups and significantly higher global efficiencies were found in the networks of the latter group. Moreover, we showed significant correlations between IQ scores and network properties across all subjects while controlling for age and gender. Specifically, higher intelligence scores corresponded to a shorter characteristic path length and a higher global efficiency of the networks, indicating a more efficient parallel information transfer in the brain. The results were consistently observed not only in the binary but also in the weighted networks, which together provide convergent evidence for our hypothesis. Our findings suggest that the efficiency of brain structural organization may be an important biological basis for intelligence.
Link
May 28, 2009
Ancient mtDNA from Pompeii
From the paper:
Annals of Human Genetics doi:10.1111/j.1469-1809.2009.00520.x
Ancient DNA and Family Relationships in a Pompeian House
Giovanni Di Bernardo et al.
Abstract
Archaeological, anthropological and pathological data suggest that thirteen skeletons found in a house at the Pompeii archaeological site, dated to 79 A.D., belong to one family. To verify this and to identify the relationships between these individuals, we analyzed DNA extracted from bone specimens. Specifically, hypervariable segment 1 (HVS1) of the human mitochondrial DNA (mtDNA) control region was amplified in two overlapping polymerase chain reactions and the sequences were compared to the revised Cambridge Reference Sequence. As independent controls, other polymorphic sites in HVS1, HVS2 and in the coding region were analyzed. We also amplified some short tandem repeats of the thirteen specimens. This study revealed that six of the thirteen individuals are indeed closely related.
Link
The aim of this study was to investigate family relationships among the inhabitants of Cajus Iulius Polybius’ house. In addition, the mtDNA haplogroup attribution of these individuals was considered.Some pictures from the house of Polybius.
...
Regarding the mtDNA haplogroups, individuals 3A, 3B, 2A, 2B, 2C, 3D most likely belong to haplogroup T2b (Malyarchuk & Derenko, 1999; Pike, 2006; Richards et al., 2000), which in modern Italians ranges between 4.4 and 4.7% (A. Torroni, personal communication). The polymorphisms in positions 16294 and 16304 detected in individuals 3A, 3B, 2A, 2B, 2C, 3D in haplogroup T2b are often associated with the polymorphism at nucleotide 16296 (Pike, 2006; Richards et al., 2000). The absence of this mutation in our individuals is probably due to the instability of this nucleotide in haplogroup T2b (Malyarchuk & Derenko, 1999; Richards et al., 2000; Pike, 2006).
...
Individual 1A, besides the polymorphism at position 16399 in HVS1, shows a sequence identical to rCRS at position 7028 hence we might assume that he most likely belongs to haplogroup H (Torroni et al., 1996). Individual 1B, showing polymorphisms at positions 16292 and 16298 and a sequence identical to rCRS at positions 73 and 4580, most likely belongs to haplogroup HV0 (Achilli et al., 2007; Pierron et al., 2008).
...
For individual 5,6A, who shows a polymorphism at positions 16391 and 73, and for 5,6B who shares the rCRS in HVS1 and at position 73, we can conclude that while individual 5,6A cannot be attributed to haplogroup H, individual 5,6B most likely can be (Torroni et al., 1996).
Annals of Human Genetics doi:10.1111/j.1469-1809.2009.00520.x
Ancient DNA and Family Relationships in a Pompeian House
Giovanni Di Bernardo et al.
Abstract
Archaeological, anthropological and pathological data suggest that thirteen skeletons found in a house at the Pompeii archaeological site, dated to 79 A.D., belong to one family. To verify this and to identify the relationships between these individuals, we analyzed DNA extracted from bone specimens. Specifically, hypervariable segment 1 (HVS1) of the human mitochondrial DNA (mtDNA) control region was amplified in two overlapping polymerase chain reactions and the sequences were compared to the revised Cambridge Reference Sequence. As independent controls, other polymorphic sites in HVS1, HVS2 and in the coding region were analyzed. We also amplified some short tandem repeats of the thirteen specimens. This study revealed that six of the thirteen individuals are indeed closely related.
Link
Heterozygosity and heart-related quantitative traits
Annals of Human Genetics doi:10.1111/j.1469-1809.2009.00514.x
Association between SNP Heterozygosity and Quantitative Traits in the Framingham Heart Study
Didahally R. Govindaraju et al.
Abstract
Associations between multilocus heterozygosity and fitness traits, also termed heterozygosity and fitness correlations (HFCs), have been reported in numerous organisms. These studies, in general, indicate a positive relationship between heterozygosity and fitness traits. We studied the association between genome-wide heterozygosity at 706 non-synonymous and synonymous SNPs and 19 quantitative traits, including morphological, biochemical and fitness traits in the Framingham Heart Study. Statistically significant association was found between heterozygosity and systolic and diastolic blood pressures as well as left ventricular diameter and wall thickness. These results suggest that heterozygosity may be associated with traits, such as blood pressure that closely track environmental variations. Balancing selection may be operating in the maintenance of heterozygosity and the major components of blood pressure and hypertension. Genome wide SNP heterozygosity may be used to understand the phenomenon of dominance as well as the evolutionary basis of many quantitative traits in humans.
Link
Association between SNP Heterozygosity and Quantitative Traits in the Framingham Heart Study
Didahally R. Govindaraju et al.
Abstract
Associations between multilocus heterozygosity and fitness traits, also termed heterozygosity and fitness correlations (HFCs), have been reported in numerous organisms. These studies, in general, indicate a positive relationship between heterozygosity and fitness traits. We studied the association between genome-wide heterozygosity at 706 non-synonymous and synonymous SNPs and 19 quantitative traits, including morphological, biochemical and fitness traits in the Framingham Heart Study. Statistically significant association was found between heterozygosity and systolic and diastolic blood pressures as well as left ventricular diameter and wall thickness. These results suggest that heterozygosity may be associated with traits, such as blood pressure that closely track environmental variations. Balancing selection may be operating in the maintenance of heterozygosity and the major components of blood pressure and hypertension. Genome wide SNP heterozygosity may be used to understand the phenomenon of dominance as well as the evolutionary basis of many quantitative traits in humans.
Link
May 27, 2009
The first Korean genome
An interesting bit:
So -based on indels- the Korean and Chinese individuals are ~24 times less distant to each other than the Korean is to James Watson (a European descendant) and ~13 times less distant to each other than the Korean was to NA18507 (a Nigerian). Table 4 in the paper has all the detailed numbers.
Figure 2 shows the overlap -number of SNPs- between various full genomes available.

Consider (E): 1.2 million SNPs are shared by the Korean and Venter/Watson; ~0.5 million are shared by the Korean and Venter (but not Watson) and the Korean and Watson (but not Venter), i.e., they transcend racial lines.
But, another ~0.5 million is shared by Venter and Watson, but not the Korean. A subset of these may be shared by accident for these three individuals (i.e., another Korean might also possess some of them). Another subset may be shared by Venter and Watson and most other Caucasoids; another subset may be shared by Venter and Watson, presumably due to their common Western European ancestry (or shared other minor ancestry), and so on.
As we sample more full genomes, we will be able to zero in on the pan-human SNPs, which represent shared human genetic diversity, as well as SNPs limited to races, subraces, ethnic groups, regions, ..., individuals.
This is an open access paper, so you can read it for yourselves.
Genome Research doi:10.1101/gr.092197.109
The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group
Sung-Min Ahn et al.
Abstract
In addition, the comparison of indels between SJK and YH (Table 4) showed that the two genomes shared the same type of indels by 99.5% on the same genomic loci (SJK and HuRef shared 86.2%, SJK and Watson shared 87.8%, SJK and NA18507 shared 93.6%).
So -based on indels- the Korean and Chinese individuals are ~24 times less distant to each other than the Korean is to James Watson (a European descendant) and ~13 times less distant to each other than the Korean was to NA18507 (a Nigerian). Table 4 in the paper has all the detailed numbers.
Figure 2 shows the overlap -number of SNPs- between various full genomes available.

Consider (E): 1.2 million SNPs are shared by the Korean and Venter/Watson; ~0.5 million are shared by the Korean and Venter (but not Watson) and the Korean and Watson (but not Venter), i.e., they transcend racial lines.
But, another ~0.5 million is shared by Venter and Watson, but not the Korean. A subset of these may be shared by accident for these three individuals (i.e., another Korean might also possess some of them). Another subset may be shared by Venter and Watson and most other Caucasoids; another subset may be shared by Venter and Watson, presumably due to their common Western European ancestry (or shared other minor ancestry), and so on.
As we sample more full genomes, we will be able to zero in on the pan-human SNPs, which represent shared human genetic diversity, as well as SNPs limited to races, subraces, ethnic groups, regions, ..., individuals.
This is an open access paper, so you can read it for yourselves.
Genome Research doi:10.1101/gr.092197.109
The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group
Sung-Min Ahn et al.
Abstract
We present the first Korean individual genome sequence (SJK) and analysis results. The diploid genome of a Korean male was sequenced to 28.95-fold redundancy using the Illumina paired-end sequencing method. SJK covered 99.9% of the NCBI human reference genome. We identified 420,083 novel SNPs that are not in the dbSNP database. Despite a close similarity, significant differences were observed between the Chinese genome (YH), the only other Asian genome available, and SJK: 1) 39.87% (1,371,239 out of 3,439,107) SNPs were SJK-specific (49.51% against Venter's, 46.94% against Watson's, and 44.17% against the Yoruba genomes), 2) 99.5% (22,495 out of 22,605) of short indels (less than 4 bp) discovered on the same loci had the same size and type as YH, and 3) 11.3% (331 out of 2920) deletion structural variants were SJK-specific. Even after attempting to map unmapped reads of SJK to unanchored NCBI scaffolds, HGSV, and available personal genomes, there were still 5.77% SJK reads that could not be mapped. All these findings indicate that the overall genetic differences among individuals from closely related ethnic groups may be significant. Hence, constructing reference genomes for minor socio-ethnic groups will be useful for massive individual genome sequencing.
Evidence of leprosy in India (~2,000BC)
Anthropology.net points me to this new paper. I recommend that post for more details on the study.
PLoS ONE doi:10.1371/journal.pone.0005669
Ancient Skeletal Evidence for Leprosy in India (2000 B.C.)
Gwen Robbins et al.
Abstract
PLoS ONE doi:10.1371/journal.pone.0005669
Ancient Skeletal Evidence for Leprosy in India (2000 B.C.)
Gwen Robbins et al.
Abstract
Background
Leprosy is a chronic infectious disease caused by Mycobacterium leprae that affects almost 250,000 people worldwide. The timing of first infection, geographic origin, and pattern of transmission of the disease are still under investigation. Comparative genomics research has suggested M. leprae evolved either in East Africa or South Asia during the Late Pleistocene before spreading to Europe and the rest of the World. The earliest widely accepted evidence for leprosy is in Asian texts dated to 600 B.C.
Methodology/Principal Findings
We report an analysis of pathological conditions in skeletal remains from the second millennium B.C. in India. A middle aged adult male skeleton demonstrates pathological changes in the rhinomaxillary region, degenerative joint disease, infectious involvement of the tibia (periostitis), and injury to the peripheral skeleton. The presence and patterning of lesions was subject to a process of differential diagnosis for leprosy including treponemal disease, leishmaniasis, tuberculosis, osteomyelitis, and non-specific infection.
Conclusions/Significance
Results indicate that lepromatous leprosy was present in India by 2000 B.C. This evidence represents the oldest documented skeletal evidence for the disease. Our results indicate that Vedic burial traditions in cases of leprosy were present in northwest India prior to the first millennium B.C. Our results also support translations of early Vedic scriptures as the first textual reference to leprosy. The presence of leprosy in skeletal material dated to the post-urban phase of the Indus Age suggests that if M. leprae evolved in Africa, the disease migrated to India before the Late Holocene, possibly during the third millennium B.C. at a time when there was substantial interaction among the Indus Civilization, Mesopotamia, and Egypt. This evidence should be impetus to look for additional skeletal and molecular evidence of leprosy in India and Africa to confirm the African origin of the disease.
LinkMay 25, 2009
MHC-dissimilar mating in Brazil
This seems to parallel previous findings on European Americans.
Opposites attract -- how genetics influences humans to choose their mates
I had previously posted some more abstracts from ESHG 2009. Here is the abstract from this study:
New evidences about MHC-based patterns of mate choice
Opposites attract -- how genetics influences humans to choose their mates
Vienna, Austria: New light has been thrown on how humans choose their partners, a scientist will tell the annual conference of the European Society of Human Genetics today (Monday May 25). Professor Maria da Graça Bicalho, head of the Immunogenetics and Histocompatibility Laboratory at the University of Parana, Brazil, says that her research had shown that people with diverse major histocompatibility complexes (MHCs) were more likely to choose each other as mates than those whose MHCs were similar, and that this was likely to be an evolutionary strategy to ensure healthy reproduction.
Females' preference for MHC dissimilar mates has been shown in many vertebrate species, including humans, and it is also known that MHC influences mating selection by preferences for particular body odours. The Brazilian team has been working in this field since 1998, and decided to investigate mate selection in the Brazilian population, while trying to uncover the biological significance of MHC diversity.
The scientists studied MHC data from 90 married couples, and compared them with 152 randomly-generated control couples. They counted the number of MHC dissimilarities among those who were real couples, and compared them with those in the randomly-generated 'virtual couples'. "If MHC genes did not influence mate selection", says Professor Bicalho, "we would have expected to see similar results from both sets of couples. But we found that the real partners had significantly more MHC dissimilarities than we could have expected to find simply by chance."
Within MHC-dissimilar couples the partners will be genetically different, and such a pattern of mate choice decreases the danger of endogamy (mating among relatives) and increases the genetic variability of offspring. Genetic variability is known to be an advantage for offspring, and the MHC effect could be an evolutionary strategy underlying incest avoidance in humans and also improving the efficiency of the immune system, the scientists say.
The MHC is a large genetic region situated on chromosome 6, and found in most vertebrates. It plays an important role in the immune system and also in reproductive success. Apart from being a large region, it is also an extraordinarily diverse one.
"Although it may be tempting to think that humans choose their partners because of their similarities", says Professor Bicalho, "our research has shown clearly that it is differences that make for successful reproduction, and that the subconscious drive to have healthy children is important when choosing a mate."
The scientists believe that their findings will help understanding of conception, fertility, and gestational failures. Research has already shown that couples with similar MHC genes had longer intervals between births, which could imply early, unperceived miscarriages. "We intend to follow up this work by looking at social and cultural influences as well as biological ones in mate choice, and relating these to the genetic diversity of the extended MHC region", says Professor Bicalho.
"We expect to find that cultural aspects play an important role in mate choice, and certainly do not subscribe to the theory that if a person bears a particular genetic variant it will determine his or her behaviour. But we also think that the unconscious evolutionary aspect of partner choice should not be overlooked. We believe our research shows that this has an important role to play in ensuring healthy reproduction, by helping to ensure that children are born with a strong immune system better able to cope with infection."
I had previously posted some more abstracts from ESHG 2009. Here is the abstract from this study:
New evidences about MHC-based patterns of mate choice
M. Bicalho, J. da Silva, J. M. Magalhães, W. Silva;
Major Histocompatibility Complex (MHC) genes code for cell surface proteins, which plays an important role in immune recognition. In the late 1970s, Yamazaki observed that inbred mice were more likely to mate with partners having MHC dissimilar genes. Females’ preference for MHC dissimilar mates was also observed in other vertebrate species, including humans. It has been shown that MHC influences mating selection mediated by preferences based on body odor. What’s the functional significance of these findings, if some? It was assumed that through olfactory cues MHC-related evolved as a strategy to maximize the offspring MHC heterozygosity. Parents with dissimilar MHCs could provide their offspring with a better chance to ward infections off because their immune system genes are more diverse. MHC genotype might be used to signal relatedness and immune response genotypes through.
We investigated whether husband-wife couples (n=90) obtained from LIGH’s database were more MHC-similar/dissimilar in comparison to random couples generated from the same database (n=55 000) as to collect evidence of MHC influence in MHC-based patterns of mate choice.
The individuals HLA typing (Class I and Class II) was performed by PCR-SSP or PCR-rSSOP using a commercial kit ( One Lambda Inc., Canoga Park. CA, USA).
Our results and comparisons ( p= 0,014) suggest that couples seem to be formed by individuals with less HLA similarity, corroborating the hypothesis that HLA antigens, especially Class I, may influence mate selection and marriages in humans.
May 24, 2009
European admixture and Body Mass Index in African Americans
While a larger portion of African ancestry in African Americans is associated with higher probability of obesity, a larger portion of African ancestry in a particular locus actually reduced the probability of obesity. This underscores the importance of not relying on first-order approximations (racial identity) when more detailed information is available.
From the paper:
Admixture Mapping of 15,280 African Americans Identifies Obesity Susceptibility Loci on Chromosomes 5 and X
Ching-Yu Cheng et al.
Abstract
The prevalence of obesity (body mass index (BMI) ≥30 kg/m2) is higher in African Americans than in European Americans, even after adjustment for socioeconomic factors, suggesting that genetic factors may explain some of the difference. To identify genetic loci influencing BMI, we carried out a pooled analysis of genome-wide admixture mapping scans in 15,280 African Americans from 14 epidemiologic studies. Samples were genotyped at a median of 1,411 ancestry-informative markers. After adjusting for age, sex, and study, BMI was analyzed both as a dichotomized (top 20% versus bottom 20%) and a continuous trait. We found that a higher percentage of European ancestry was significantly correlated with lower BMI (ρ = −0.042, P = 1.6×10−7). In the dichotomized analysis, we detected two loci on chromosome X as associated with increased African ancestry: the first at Xq25 (locus-specific LOD = 5.94; genome-wide score = 3.22; case-control Z = −3.94); and the second at Xq13.1 (locus-specific LOD = 2.22; case-control Z = −4.62). Quantitative analysis identified a third locus at 5q13.3 where higher BMI was highly significantly associated with greater European ancestry (locus-specific LOD = 6.27; genome-wide score = 3.46). Further mapping studies with dense sets of markers will be necessary to identify the alleles in these regions of chromosomes X and 5 that may be associated with variation in BMI.
Link
From the paper:
We have carried out admixture mapping analyses to search for genomic regions associated with BMI. This pooled analysis of samples from 14 studies is the largest admixture scan reported to date. In more than 15,000 individuals, we identified a locus on chromosome 5 where greater local European ancestry was associated with higher levels of BMI (P = 5.8×10−7), and two regions on chromosome X where greater local European ancestry was associated with lower levels of BMI (both P<5.0×10−6). Each of these three associations was above and beyond the contribution of genome-wide European ancestry, and each reached genome-wide significance.PLoS Genetics doi:10.1371/journal.pgen.1000490
...
The inverse correlation between BMI and percentage of European ancestry estimated on the genome-wide scale confirmed the results from previous studies of smaller sample size and fewer markers [29],[30]. However, while genome-wide ancestry is likely correlated with local ancestry, it cannot fully capture ancestry information at each locus as there exists variation across the genome in the effects of locus-specific ancestry on obesity. In particular, local European ancestry at 5q13.3 was positively associated with BMI, providing the first evidence of a genome-wide significant ancestry association being in the opposite direction to the overall epidemiological association.
Admixture Mapping of 15,280 African Americans Identifies Obesity Susceptibility Loci on Chromosomes 5 and X
Ching-Yu Cheng et al.
Abstract
The prevalence of obesity (body mass index (BMI) ≥30 kg/m2) is higher in African Americans than in European Americans, even after adjustment for socioeconomic factors, suggesting that genetic factors may explain some of the difference. To identify genetic loci influencing BMI, we carried out a pooled analysis of genome-wide admixture mapping scans in 15,280 African Americans from 14 epidemiologic studies. Samples were genotyped at a median of 1,411 ancestry-informative markers. After adjusting for age, sex, and study, BMI was analyzed both as a dichotomized (top 20% versus bottom 20%) and a continuous trait. We found that a higher percentage of European ancestry was significantly correlated with lower BMI (ρ = −0.042, P = 1.6×10−7). In the dichotomized analysis, we detected two loci on chromosome X as associated with increased African ancestry: the first at Xq25 (locus-specific LOD = 5.94; genome-wide score = 3.22; case-control Z = −3.94); and the second at Xq13.1 (locus-specific LOD = 2.22; case-control Z = −4.62). Quantitative analysis identified a third locus at 5q13.3 where higher BMI was highly significantly associated with greater European ancestry (locus-specific LOD = 6.27; genome-wide score = 3.46). Further mapping studies with dense sets of markers will be necessary to identify the alleles in these regions of chromosomes X and 5 that may be associated with variation in BMI.
Link
May 23, 2009
Y chromosome population structure in Arabian peninsula
 On the left, the MDS plot of genetic distances in studied populations and others from the literature. The haplotypes are available in free supplementary material (pdf). Someone ought to feed these to Whit Athey's haplogroup predictor to get estimates of the haplogroup composition of the Arabian populations.
On the left, the MDS plot of genetic distances in studied populations and others from the literature. The haplotypes are available in free supplementary material (pdf). Someone ought to feed these to Whit Athey's haplogroup predictor to get estimates of the haplogroup composition of the Arabian populations.Hum Hered 2009;68:45-54 (DOI: 10.1159/000210448)
Local Population Structure in Arabian Peninsula Revealed by Y-STR Diversity
Farida Alshamali et al.
Abstract
Genetic studies have been underway on Arabian Peninsula populations because of their pivotal geographic location for population migration and times of occurrence. To assist in better understanding population dynamics in this region, evidence is presented herein on local population structure in the Arabian Peninsula, based on Y-STR characterisation in four Arabian samples and its comparison in a broad geographical scale. Our results demonstrate that geography played an important role in shaping the genetic structure of the region around the Near-East. Populations are grouped regionally but none of these groups is significantly differentiated from others and all groups merge in the Near-East, in keeping with this important migration corridor for the human species. Focusing on the Arabian Peninsula, we show that Dubai and Oman share genetic affinities with other Near-Eastern populations, while Saudi Arabia and Yemen show a relative distinctive isolated background. Those two populations may have been kept relatively separated from migration routes, maybe due to their location in a desert area.
Link
May 22, 2009
Macedonia Evidence initiative
A very worthwhile effort. From the About section of the website:
The text of the letter:Classical Scholars from around the world, well known for their expertise in the history of Greece are presenting, examining, and discussing the historical evidence.
The first few scholars were motivated by the article in the Archaeology magazine, and the letter Stephen G. Miller, Ph.D sent in response, which Archaeology did not publish.
Since then, the list of scholars that have examined the evidence has been growing and 222 scholars have undersigned the letter to President Barak Obama.
If you want to contribute to the discussion, please contact us at: SavingAlexander@macedonia-evidence.org.
The Frequently Asked Questions are worth reading too.Dear President Obama,
We, the undersigned scholars of Graeco-Roman antiquity, respectfully request that you intervene to clean up some of the historical debris left in southeast Europe by the previous U.S. administration.
On November 4, 2004, two days after the re-election of President George W. Bush, his administration unilaterally recognized the “Republic of Macedonia.” This action not only abrogated geographic and historic fact, but it also has unleashed a dangerous epidemic of historical revisionism, of which the most obvious symptom is the misappropriation by the government in Skopje of the most famous of Macedonians, Alexander the Great.
We believe that this silliness has gone too far, and that the U.S.A. has no business in supporting the subversion of history. Let us review facts. (The documentation for these facts [here in boldface] can be found attached and at: http://macedonia-evidence.org/documentation.html)
The land in question, with its modern capital at Skopje, was called Paionia in antiquity. Mts. Barnous and Orbelos (which form today the northern limits of Greece) provide a natural barrier that separated, and separates, Macedonia from its northern neighbor. The only real connection is along the Axios/Vardar River and even this valley “does not form a line of communication because it is divided by gorges.”
While it is true that the Paionians were subdued by Philip II, father of Alexander, in 358 B.C. they were not Macedonians and did not live in Macedonia. Likewise, for example, the Egyptians, who were subdued by Alexander, may have been ruled by Macedonians, including the famous Cleopatra, but they were never Macedonians themselves, and Egypt was never called Macedonia.
Rather, Macedonia and Macedonian Greeks have been located for at least 2,500 years just where the modern Greek province of Macedonia is. Exactly this same relationship is true for Attica and Athenian Greeks, Argos and Argive Greeks, Corinth and Corinthian Greeks, etc.
We do not understand how the modern inhabitants of ancient Paionia, who speak Slavic – a language introduced into the Balkans about a millennium after the death of Alexander – can claim him as their national hero. Alexander the Great was thoroughly and indisputably Greek. His great-great-great grandfather, Alexander I, competed in the Olympic Games where participation was limited to Greeks.
Even before Alexander I, the Macedonians traced their ancestry to Argos, and many of their kings used the head of Herakles - the quintessential Greek hero - on their coins.
Euripides – who died and was buried in Macedonia– wrote his play Archelaos in honor of the great-uncle of Alexander, and in Greek. While in Macedonia, Euripides also wrote the Bacchai, again in Greek. Presumably the Macedonian audience could understand what he wrote and what they heard.
Alexander’s father, Philip, won several equestrian victories at Olympia and Delphi, the two most Hellenic of all the sanctuaries in ancient Greece where non-Greeks were not allowed to compete. Even more significantly, Philip was appointed to conduct the Pythian Games at Delphi in 346 B.C. In other words, Alexander the Great’s father and his ancestors were thoroughly Greek. Greek was the language used by Demosthenes and his delegation from Athens when they paid visits to Philip, also in 346 B.C. Another northern Greek, Aristotle, went off to study for nearly 20 years in the Academy of Plato. Aristotle subsequently returned to Macedonia and became the tutor of Alexander III. They used Greek in their classroom which can still be seen near Naoussa in Macedonia.
Alexander carried with him throughout his conquests Aristotle’s edition of Homer’s Iliad. Alexander also spread Greek language and culture throughout his empire, founding cities and establishing centers of learning. Hence inscriptions concerning such typical Greek institutions as the gymnasium are found as far away as Afghanistan. They are all written in Greek.
The questions follow: Why was Greek the lingua franca all over Alexander’s empire if he was a “Macedonian”? Why was the New Testament, for example, written in Greek?
The answers are clear: Alexander the Great was Greek, not Slavic, and Slavs and their language were nowhere near Alexander or his homeland until 1000 years later. This brings us back to the geographic area known in antiquity as Paionia. Why would the people who live there now call themselves Macedonians and their land Macedonia? Why would they abduct a completely Greek figure and make him their national hero?
The ancient Paionians may or may not have been Greek, but they certainly became Greekish, and they were never Slavs. They were also not Macedonians. Ancient Paionia was a part of the Macedonian Empire. So were Ionia and Syria and Palestine and Egypt and Mesopotamia and Babylonia and Bactria and many more. They may thus have become “Macedonian” temporarily, but none was ever “Macedonia”. The theft of Philip and Alexander by a land that was never Macedonia cannot be justified.
The traditions of ancient Paionia could be adopted by the current residents of that geographical area with considerable justification. But the extension of the geographic term “Macedonia” to cover southern Yugoslavia cannot. Even in the late 19th century, this misuse implied unhealthy territorial aspirations.
The same motivation is to be seen in school maps that show the pseudo-greater Macedonia, stretching from Skopje to Mt. Olympus and labeled in Slavic. The same map and its claims are in calendars, bumper stickers, bank notes, etc., that have been circulating in the new state ever since it declared its independence from Yugoslavia in 1991. Why would a poor land-locked new state attempt such historical nonsense? Why would it brazenly mock and provoke its neighbor?
However one might like to characterize such behavior, it is clearly not a force for historical accuracy, nor for stability in the Balkans. It is sad that the United States of America has abetted and encouraged such behavior.
We call upon you, Mr. President, to help - in whatever ways you deem appropriate - the government in Skopje to understand that it cannot build a national identity at the expense of historic truth. Our common international society cannot survive when history is ignored, much less when history is fabricated.
May 21, 2009
Female mice prefer outbred males
BMC Evol Biol. 2009 May 16;9(1):104. [Epub ahead of print]
Females prefer the scent of outbred males: good-genes-as-heterozygosity?
Ilmonen P, Stundner G, Thosz M, Penn DJ.
ABSTRACT: BACKGROUND: There is increasing interest to determine the relative importance of non-additive genetic benefits as opposed to additive ones for the evolution of mating preferences and maintenance of genetic variation in sexual ornaments. The 'good-genes-as-heterozygosity' hypothesis predicts that females should prefer to mate with more heterozygous males to gain more heterozygous (and less inbred) offspring. Heterozygosity increases males' sexual ornamentation, mating success and reproduction success, yet few experiments have tested whether females are preferentially attracted to heterozygous males, and none have tested whether females' own heterozygosity influences their preferences. Outbred females might have the luxury of being more choosey, but on the other hand, inbred females might have more to gain by mating with heterozygous males. We manipulated heterozygosity in wild-derived house mice (Mus musculus musculus) through inbreeding and tested whether the females are more attracted to the scent of outbred versus inbred males, and whether females' own inbreeding status affects their preferences. We also tested whether infecting both inbred and outbred males with Salmonella would magnify females' preferences for outbred males. RESULTS: Females showed a significant preference for outbred males, and this preference was slightly more pronounced among inbred females. We found no evidence that Salmonella infection increased the relative attractiveness of outbred versus inbred males; however, we found no evidence that inbreeding affected males' disease resistance in this study. CONCLUSIONS: Our findings support the idea that females are more attracted to outbred males, and they suggest that such preferences may be stronger among inbred than outbred females, which is consistent with the 'good-genes-as-heterozygosity' hypothesis. It is unclear whether this odour preference reflects females' actual mating preferences, though it suggests that future studies should consider females' as well as males' heterozygosity. Our study has implications for efforts to understand how mate choice can provide genetic benefits without eroding genetic diversity (lek paradox), and also conservation efforts to determine the fitness consequences of inbreeding and the maintenance of genetic diversity in small, inbred populations.
Link
Females prefer the scent of outbred males: good-genes-as-heterozygosity?
Ilmonen P, Stundner G, Thosz M, Penn DJ.
ABSTRACT: BACKGROUND: There is increasing interest to determine the relative importance of non-additive genetic benefits as opposed to additive ones for the evolution of mating preferences and maintenance of genetic variation in sexual ornaments. The 'good-genes-as-heterozygosity' hypothesis predicts that females should prefer to mate with more heterozygous males to gain more heterozygous (and less inbred) offspring. Heterozygosity increases males' sexual ornamentation, mating success and reproduction success, yet few experiments have tested whether females are preferentially attracted to heterozygous males, and none have tested whether females' own heterozygosity influences their preferences. Outbred females might have the luxury of being more choosey, but on the other hand, inbred females might have more to gain by mating with heterozygous males. We manipulated heterozygosity in wild-derived house mice (Mus musculus musculus) through inbreeding and tested whether the females are more attracted to the scent of outbred versus inbred males, and whether females' own inbreeding status affects their preferences. We also tested whether infecting both inbred and outbred males with Salmonella would magnify females' preferences for outbred males. RESULTS: Females showed a significant preference for outbred males, and this preference was slightly more pronounced among inbred females. We found no evidence that Salmonella infection increased the relative attractiveness of outbred versus inbred males; however, we found no evidence that inbreeding affected males' disease resistance in this study. CONCLUSIONS: Our findings support the idea that females are more attracted to outbred males, and they suggest that such preferences may be stronger among inbred than outbred females, which is consistent with the 'good-genes-as-heterozygosity' hypothesis. It is unclear whether this odour preference reflects females' actual mating preferences, though it suggests that future studies should consider females' as well as males' heterozygosity. Our study has implications for efforts to understand how mate choice can provide genetic benefits without eroding genetic diversity (lek paradox), and also conservation efforts to determine the fitness consequences of inbreeding and the maintenance of genetic diversity in small, inbred populations.
Link
May 20, 2009
Review paper on Y-chromosome haplogroup E-M35 (Lancaster 2009)
This is quite useful as a reference for those interested in E-M35 and its many subclades.
A quick comment on p. 53 where Sicily is discussed and the prevalence of R1b and I1 in the West is mentioned in the context of Phoenicians who settled in West Sicily.
Haplogroup I1 is probably to a large degree due to the Normans whose capital was in Palermo (NW Sicily). R1b on the other hand may have been added by the Normans, but may also be due to the pre-Greek populations of Sicily, such as the Sicani who were (after Herodotus) of Iberian origin.
Journal of Genetic Genealogy Volume 5, Number 1, Spring, 2009
Y Haplogroups, Archaeological Cultures and Language Families: A Review of the Possibility of Multidisciplinary Comparisons Using the Case of Haplogroup E-M35
Andrew Lancaster
Abstract
Archaeology, comparative linguistics and population genetics all have something to add to speculation about early human migrations, and the three disciplines often make reference to each other in broad terms. But, in reality, the results are often disappointingly indecisive. This article explores the case of Y haplogroup E-M35 (E1b1b1), which has so far mainly only been mentioned in a passing way in archaeological and linguistic debate, but which, it shall be shown, shows great promise as more detailed about it's phylogenetic structure, and its regional distribution becomes available each year.
Link (pdf)
A quick comment on p. 53 where Sicily is discussed and the prevalence of R1b and I1 in the West is mentioned in the context of Phoenicians who settled in West Sicily.
Haplogroup I1 is probably to a large degree due to the Normans whose capital was in Palermo (NW Sicily). R1b on the other hand may have been added by the Normans, but may also be due to the pre-Greek populations of Sicily, such as the Sicani who were (after Herodotus) of Iberian origin.
Journal of Genetic Genealogy Volume 5, Number 1, Spring, 2009
Y Haplogroups, Archaeological Cultures and Language Families: A Review of the Possibility of Multidisciplinary Comparisons Using the Case of Haplogroup E-M35
Andrew Lancaster
Abstract
Archaeology, comparative linguistics and population genetics all have something to add to speculation about early human migrations, and the three disciplines often make reference to each other in broad terms. But, in reality, the results are often disappointingly indecisive. This article explores the case of Y haplogroup E-M35 (E1b1b1), which has so far mainly only been mentioned in a passing way in archaeological and linguistic debate, but which, it shall be shown, shows great promise as more detailed about it's phylogenetic structure, and its regional distribution becomes available each year.
Link (pdf)
mtDNA of Libyan Tuaregs
The sample consisted of haplogroups H1, V, M1, and an assortment of African L subclades.
From the paper:
Annals of Human Genetics doi:10.1111/j.1469-1809.2009.00526.x
First Genetic Insight into Libyan Tuaregs: A Maternal Perspective
Claudio Ottoni et al.
Abstract
The Tuaregs are a semi-nomadic pastoralist people of northwest Africa. Their origins are still a matter of debate due to the scarcity of genetic and historical data. Here we report the first data on the mitochondrial DNA (mtDNA) genetic characterization of a Tuareg sample from Fezzan (Libyan Sahara). A total of 129 individuals from two villages in the Acacus region were genetically analysed. Both the hypervariable regions and the coding region of mtDNA were investigated. Phylogeographic investigation was carried out in order to reconstruct human migratory shifts in central Sahara, and to shed light on the origin of the Libyan Tuaregs. Our results clearly show low genetic diversity in the sample, possibly due to genetic drift and founder effect associated with the separation of Libyan Tuaregs from an ancestral population. Furthermore, the maternal genetic pool of the Libyan Tuaregs is characterized by a major "European" component shared with the Berbers that could be traced to the Iberian Peninsula, as well as a minor 'south Saharan' contribution possibly linked to both Eastern African and Near Eastern populations.
Link
From the paper:
Of note is that the other Tuareg sample described in the literature (Watson et al., 1996) (Western Tuaregs) did not show a close genetic relationship with the Libyan Tuaregs, implying a genetic heterogeneity of the Tuaregs. This difference appears to be primarily caused by the low frequency (8%) of the European component in the Western Tuaregs, characteristic of northern African populations. After the removal of the H and V haplotypes, the Libyan Tuaregs showed a strong affiliation with the Eastern populations, while theWestern Tuaregs associated more with the Central and Western African populations (Fig. 2).In other words, the Libyan Tuaregs are primarily "European"+"Eastern African" (or Saharan) in affiliation, while the Western Tuaregs from Nigeria, Niger, and Mali have a predominantly West African maternal heritage.
Annals of Human Genetics doi:10.1111/j.1469-1809.2009.00526.x
First Genetic Insight into Libyan Tuaregs: A Maternal Perspective
Claudio Ottoni et al.
Abstract
The Tuaregs are a semi-nomadic pastoralist people of northwest Africa. Their origins are still a matter of debate due to the scarcity of genetic and historical data. Here we report the first data on the mitochondrial DNA (mtDNA) genetic characterization of a Tuareg sample from Fezzan (Libyan Sahara). A total of 129 individuals from two villages in the Acacus region were genetically analysed. Both the hypervariable regions and the coding region of mtDNA were investigated. Phylogeographic investigation was carried out in order to reconstruct human migratory shifts in central Sahara, and to shed light on the origin of the Libyan Tuaregs. Our results clearly show low genetic diversity in the sample, possibly due to genetic drift and founder effect associated with the separation of Libyan Tuaregs from an ancestral population. Furthermore, the maternal genetic pool of the Libyan Tuaregs is characterized by a major "European" component shared with the Berbers that could be traced to the Iberian Peninsula, as well as a minor 'south Saharan' contribution possibly linked to both Eastern African and Near Eastern populations.
Link
AVPR1A and Musical Aptitude
AVPR1A has been previously implicated in dance ability and pair bonding.
PLoS ONE doi:10.1371/journal.pone.0005534
Musical Aptitude Is Associated with AVPR1A-Haplotypes
Liisa T. Ukkola et al.
Abstract
Artistic creativity forms the basis of music culture and music industry. Composing, improvising and arranging music are complex creative functions of the human brain, which biological value remains unknown. We hypothesized that practicing music is social communication that needs musical aptitude and even creativity in music. In order to understand the neurobiological basis of music in human evolution and communication we analyzed polymorphisms of the arginine vasopressin receptor 1A (AVPR1A), serotonin transporter (SLC6A4), catecol-O-methyltranferase (COMT), dopamin receptor D2 (DRD2) and tyrosine hydroxylase 1 (TPH1), genes associated with social bonding and cognitive functions in 19 Finnish families (n = 343 members) with professional musicians and/or active amateurs. All family members were tested for musical aptitude using the auditory structuring ability test (Karma Music test; KMT) and Carl Seashores tests for pitch (SP) and for time (ST). Data on creativity in music (composing, improvising and/or arranging music) was surveyed using a web-based questionnaire. Here we show for the first time that creative functions in music have a strong genetic component (h2 = .84; composing h2 = .40; arranging h2 = .46; improvising h2 = .62) in Finnish multigenerational families. We also show that high music test scores are significantly associated with creative functions in music (p<.0001). We discovered an overall haplotype association with AVPR1A gene (markers RS1 and RS3) and KMT (p = 0.0008; corrected p = 0.00002), SP (p = 0.0261; corrected p = 0.0072) and combined music test scores (COMB) (p = 0.0056; corrected p = 0.0006). AVPR1A haplotype AVR+RS1 further suggested a positive association with ST (p = 0.0038; corrected p = 0.00184) and COMB (p = 0.0083; corrected p = 0.0040) using haplotype-based association test HBAT. The results suggest that the neurobiology of music perception and production is likely to be related to the pathways affecting intrinsic attachment behavior.
PLoS ONE doi:10.1371/journal.pone.0005534
Musical Aptitude Is Associated with AVPR1A-Haplotypes
Liisa T. Ukkola et al.
Abstract
Artistic creativity forms the basis of music culture and music industry. Composing, improvising and arranging music are complex creative functions of the human brain, which biological value remains unknown. We hypothesized that practicing music is social communication that needs musical aptitude and even creativity in music. In order to understand the neurobiological basis of music in human evolution and communication we analyzed polymorphisms of the arginine vasopressin receptor 1A (AVPR1A), serotonin transporter (SLC6A4), catecol-O-methyltranferase (COMT), dopamin receptor D2 (DRD2) and tyrosine hydroxylase 1 (TPH1), genes associated with social bonding and cognitive functions in 19 Finnish families (n = 343 members) with professional musicians and/or active amateurs. All family members were tested for musical aptitude using the auditory structuring ability test (Karma Music test; KMT) and Carl Seashores tests for pitch (SP) and for time (ST). Data on creativity in music (composing, improvising and/or arranging music) was surveyed using a web-based questionnaire. Here we show for the first time that creative functions in music have a strong genetic component (h2 = .84; composing h2 = .40; arranging h2 = .46; improvising h2 = .62) in Finnish multigenerational families. We also show that high music test scores are significantly associated with creative functions in music (p<.0001). We discovered an overall haplotype association with AVPR1A gene (markers RS1 and RS3) and KMT (p = 0.0008; corrected p = 0.00002), SP (p = 0.0261; corrected p = 0.0072) and combined music test scores (COMB) (p = 0.0056; corrected p = 0.0006). AVPR1A haplotype AVR+RS1 further suggested a positive association with ST (p = 0.0038; corrected p = 0.00184) and COMB (p = 0.0083; corrected p = 0.0040) using haplotype-based association test HBAT. The results suggest that the neurobiology of music perception and production is likely to be related to the pathways affecting intrinsic attachment behavior.
Origin of San Teodoro Paleolithic Sicilians
Journal of Human Evolution doi:10.1016/j.jhevol.2009.02.002
Late Pleistocene human evolution in Sicily: comparative morphometric analysis of Grotta di San Teodoro craniofacial remains
Giuseppe D'Amore et al.
Abstract
The paleoanthropological remains from Grotta di San Teodoro near Acquedolci (province of Messina, Italy) represent the oldest and largest skeletal collection yet found documenting human settlement of Sicily. The sample, attributed to the Late Epigravettian (between 14,000 and 10,000 years B.P.), consists of seven variously complete adult individuals (San Teodoro 1–7). We compare the cranial sample to an array of both prehistoric and recent samples using multivariate techniques including D2 distance analysis, canonical variate analysis, cluster analysis, and multidimensional scaling. Overall, the San Teodoro cranial sample displays a morphometric pattern close to Western European groups of similar antiquity, in particular those from Central and Southern Italy. The morphometric affinities indicate that these people probably came from peninsular Italy by sea during the Late Epigravettian epoch. An alternative hypothesis is that they descended from immigrants that arrived by land during a low sea level episode corresponding to the maximum Würmian regression, about 18,000 years B.P, with gene flow accounting for the morphological homogeneity with the populations of peninsular Italy. The San Teodoro skeletal sample provides the first reliable evidence for human settlement of Sicily.
Link
Late Pleistocene human evolution in Sicily: comparative morphometric analysis of Grotta di San Teodoro craniofacial remains
Giuseppe D'Amore et al.
Abstract
The paleoanthropological remains from Grotta di San Teodoro near Acquedolci (province of Messina, Italy) represent the oldest and largest skeletal collection yet found documenting human settlement of Sicily. The sample, attributed to the Late Epigravettian (between 14,000 and 10,000 years B.P.), consists of seven variously complete adult individuals (San Teodoro 1–7). We compare the cranial sample to an array of both prehistoric and recent samples using multivariate techniques including D2 distance analysis, canonical variate analysis, cluster analysis, and multidimensional scaling. Overall, the San Teodoro cranial sample displays a morphometric pattern close to Western European groups of similar antiquity, in particular those from Central and Southern Italy. The morphometric affinities indicate that these people probably came from peninsular Italy by sea during the Late Epigravettian epoch. An alternative hypothesis is that they descended from immigrants that arrived by land during a low sea level episode corresponding to the maximum Würmian regression, about 18,000 years B.P, with gene flow accounting for the morphological homogeneity with the populations of peninsular Italy. The San Teodoro skeletal sample provides the first reliable evidence for human settlement of Sicily.
Link
May 19, 2009
More on prehistoric South Siberians (Keyser et al. 2009)
This seems like a compendium of these authors' previous work (see here and links therein) which had appeared in forensic journals so far; there seems to be more material in this paper than in the previous shorter papers, but as far as I can tell, no new genetic results.
There is also supplementary data in the article.
From the paper:
More from the paper:
The mtDNA results:
Human Genetics doi:10.1007/s00439-009-0683-0
Ancient DNA provides new insights into the history of south Siberian Kurgan people.
Keyser C. et al.
Abstract
To help unravel some of the early Eurasian steppe migration movements, we determined the Y-chromosomal and mitochondrial haplotypes and haplogroups of 26 ancient human specimens from the Krasnoyarsk area dated from between the middle of the second millennium BC. to the fourth century AD. In order to go further in the search of the geographic origin and physical traits of these south Siberian specimens, we also typed phenotype-informative single nucleotide polymorphisms. Our autosomal, Y-chromosomal and mitochondrial DNA analyses reveal that whereas few specimens seem to be related matrilineally or patrilineally, nearly all subjects belong to haplogroup R1a1-M17 which is thought to mark the eastward migration of the early Indo-Europeans. Our results also confirm that at the Bronze and Iron Ages, south Siberia was a region of overwhelmingly predominant European settlement, suggesting an eastward migration of Kurgan people across the Russo-Kazakh steppe. Finally, our data indicate that at the Bronze and Iron Age timeframe, south Siberians were blue (or green)-eyed, fair-skinned and light-haired people and that they might have played a role in the early development of the Tarim Basin civilization. To the best of our knowledge, no equivalent molecular analysis has been undertaken so far.
Link
There is also supplementary data in the article.
From the paper:
The additional analysis performed on Xiongnu specimens revealed that whereas none of the specimens from the Egyin Gol valley bore this haplogroup, the Scytho-Siberian skeleton from the Sebÿstei site exhibited R1a1 haplogroup.A previous study on Egyin Gol from Mongolia by Keyser et al.
More from the paper:
A search in the YHRD database as well as in our own databank revealed that none of the Y-STR haplotypes obtained from the south Siberian samples perfectly matched (at 17 loci) those included in the databases. Nevertheless, when not all loci were scored, matches were found for all samples except two (S07 and S32) for which even the search based on the 9-loci minimal haplotype was fruitless (Table 4).The article includes fairly comprehensive searches of the discovered Y-chromosome and mtDNA types in modern populations.
The mtDNA results:
Twenty samples were found to belong to west Eurasian haplogroups (U2, U4,Interestingly:
U5a1, T1, T3, T4, H5a, H6, HV, K, and I), whereas the 6 remaining samples were attributed to east Eurasian haplogroups (Z, G2a, C, F1b and N9a).
Moreover, it is likely that some mtDNA lineages were carried to southern Siberia from the Volga–Ural region. Incidentally, in the fifth century BC, Herodotus mentioned transit trade occurring in Central Asia along a route that stretched from the Urals in the west to the Altai and the Minusinsk Basin in the east (Hemphill and Mallory 2004). In Altai, the presence of the R1a1 haplogroup in the middle of the fifth century BC is confirmed by the sample SEB 96K2 of Ricaut et al. (2004) which was found to belong to this Y-haplogroup. The boundary of the eastern European influence seems to be fixed at the peri-Baikal area since no R1a1 haplogroup was found in the Xiongnu specimens of the Northern border of Mongolia.Link to Ricaut et al. (2004). This is in good agreement with the anthropological picture by Alexeev:
"The boundary of the Europeoid movement is clearly fixed at Lake Baikal. To the east of Baikal no palaeoanthropological find bears any traces of Europeoid admixture."See also my compendium on ancient Y-chromosome studies.
Human Genetics doi:10.1007/s00439-009-0683-0
Ancient DNA provides new insights into the history of south Siberian Kurgan people.
Keyser C. et al.
Abstract
To help unravel some of the early Eurasian steppe migration movements, we determined the Y-chromosomal and mitochondrial haplotypes and haplogroups of 26 ancient human specimens from the Krasnoyarsk area dated from between the middle of the second millennium BC. to the fourth century AD. In order to go further in the search of the geographic origin and physical traits of these south Siberian specimens, we also typed phenotype-informative single nucleotide polymorphisms. Our autosomal, Y-chromosomal and mitochondrial DNA analyses reveal that whereas few specimens seem to be related matrilineally or patrilineally, nearly all subjects belong to haplogroup R1a1-M17 which is thought to mark the eastward migration of the early Indo-Europeans. Our results also confirm that at the Bronze and Iron Ages, south Siberia was a region of overwhelmingly predominant European settlement, suggesting an eastward migration of Kurgan people across the Russo-Kazakh steppe. Finally, our data indicate that at the Bronze and Iron Age timeframe, south Siberians were blue (or green)-eyed, fair-skinned and light-haired people and that they might have played a role in the early development of the Tarim Basin civilization. To the best of our knowledge, no equivalent molecular analysis has been undertaken so far.
Link
May 18, 2009
Review article on epigenetic inheritance
From the paper:
The Quarterly Review of Biology, June 2009, vol. 84, no. 2, DOI: 10.1086/598822
TRANSGENERATIONAL EPIGENETIC INHERITANCE: PREVALENCE, MECHANISMS, AND IMPLICATIONS FOR THE STUDY OF HEREDITY AND EVOLUTION
Eva Jablonka, Gal Raz
Abstract
This review describes new developments in the study of transgenerational epigenetic inheritance, a component of epigenetics. We start by examining the basic concepts of the field and the mechanisms that underlie epigenetic inheritance. We present a comprehensive review of transgenerational cellular epigenetic inheritance among different taxa in the form of a table, and discuss the data contained therein. The analysis of these data shows that epigenetic inheritance is ubiquitous and suggests lines of research that go beyond present approaches to the subject. We conclude by exploring some of the consequences of epigenetic inheritance for the study of evolution, while also pointing to the importance of recognizing and understanding epigenetic inheritance for practical and theoretical issues in biology.
Link
Incorporating epigenetic inheritance into evolutionary theory extends the scope of evolutionary thinking and leads to notions of heredity and evolution that incorporate development. Dobzhansky's definition of evolution as “a change in the genetic composition of populations” (1937, p.11) appears to be too narrow because it does not incorporate all sources of heritable variations. Both evolution and heredity need to be redefined. Jablonka and Lamb (2007a,b,c) suggested that evolution should be redefined as the set of processes that lead to changes in the nature and frequency of heritable types in a population, and heredity as the developmental reconstruction processes that link ancestors and descendants and lead to similarity between them. These deliberately broad redefinitions allow evolutionary possibilities denied by the “Modern Synthesis” version of evolutionary theory, which states that variations are blind, are genetic (nucleic acid‐based), and that saltational events do not significantly contribute to evolutionary change (Mayr 1982). The epigenetic perspective challenges all these assumptions, and it seems that a new extended theory, informed by developmental studies and epigenetic inheritance, and incorporating Darwinian, Lamarckian, and saltational frameworks, is going to replace the Modern Synthesis version of evolution (Jablonka and Lamb 2005, 2007c). We believe, therefore, that the impact of epigenetics and epigenetic inheritance on evolutionary theory and the philosophy of biology will be profound.Some related posts:
- Epigenetics via twin studies
- Maternal inheritance and male reproductive fitness
- Epigenetic effects of slavery in African Americans (?)
- The eclipse of the gene
The Quarterly Review of Biology, June 2009, vol. 84, no. 2, DOI: 10.1086/598822
TRANSGENERATIONAL EPIGENETIC INHERITANCE: PREVALENCE, MECHANISMS, AND IMPLICATIONS FOR THE STUDY OF HEREDITY AND EVOLUTION
Eva Jablonka, Gal Raz
Abstract
This review describes new developments in the study of transgenerational epigenetic inheritance, a component of epigenetics. We start by examining the basic concepts of the field and the mechanisms that underlie epigenetic inheritance. We present a comprehensive review of transgenerational cellular epigenetic inheritance among different taxa in the form of a table, and discuss the data contained therein. The analysis of these data shows that epigenetic inheritance is ubiquitous and suggests lines of research that go beyond present approaches to the subject. We conclude by exploring some of the consequences of epigenetic inheritance for the study of evolution, while also pointing to the importance of recognizing and understanding epigenetic inheritance for practical and theoretical issues in biology.
Link
May 15, 2009
Fine-scale population structure in humans (Biswas et al. 2009)
This paper extends previous genome-wide studies which found structure within continental (mainly European) populations, by showing that PCA shows structure in all continental groups, and in more than the first two principal components that usually correspond with geography.
An interesting table from the paper shows the number of significant principal components within each continent:
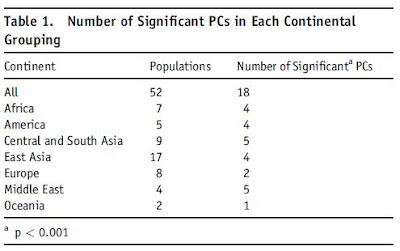
Notice the large number of principal components in the Middle East and Central Asia, suggesting a very significant differentiation in these regions, despite the small number of tested populations, and underscoring the need for more comprehensive sampling.
In the case of the Middle East, where only Afroasiatic populations were sampled, this is even more remarkable; further study of Indo-European, Turkic, and Caucasian speakers from the region will no doubt reveal further differences among them. As for Central Asia, the large number of significant principal components is related to both the inter-racial difference between Caucasoids and Mongoloids in this contact zone, as well as intra-racial differences.
The supplementary material (pdfs) has plots for the significant components of the seven continental regions. Progress will now occur by extending population sampling to unexamined populations and by full-genome sequencing which will allow us to detect finer-level distinctions even in fairly homogeneous regions of the world such as Europe.
American Journal of Human Genetics doi:10.1016/j.ajhg.2009.04.015
Genome-wide Insights into the Patterns and Determinants of Fine-Scale Population Structure in Humans
Shameek Biswas et al.
Abstract
Studying genomic patterns of human population structure provides important insights into human evolutionary history and the relationship among populations, and it has significant practical implications for disease-gene mapping. Here we describe a principal component (PC)-based approach to studying intracontinental population structure in humans, identify the underlying markers mediating the observed patterns of fine-scale population structure, and infer the predominating evolutionary forces shaping local population structure. We applied this methodology to a data set of 650K SNPs genotyped in 944 unrelated individuals from 52 populations and demonstrate that, although typical PC analyses focus on the top axes of variation, substantial information about population structure is contained in lower-ranked PCs. We identified 18 significant PCs, some of which distinguish individual populations. In addition to visually representing sample clusters in PC biplots, we estimated the set of all SNPs significantly correlated with each of the most informative axes of variation. These polymorphisms, unlike ancestry-informative markers (AIMs), constitute a much larger set of loci that drive genomic signatures of population structure. The genome-wide distribution of these significantly correlated markers can largely be accounted for by the stochastic effects of genetic drift, although significant clustering does occur in genomic regions that have been previously implicated as targets of recent adaptive evolution.
Link
An interesting table from the paper shows the number of significant principal components within each continent:

Notice the large number of principal components in the Middle East and Central Asia, suggesting a very significant differentiation in these regions, despite the small number of tested populations, and underscoring the need for more comprehensive sampling.
In the case of the Middle East, where only Afroasiatic populations were sampled, this is even more remarkable; further study of Indo-European, Turkic, and Caucasian speakers from the region will no doubt reveal further differences among them. As for Central Asia, the large number of significant principal components is related to both the inter-racial difference between Caucasoids and Mongoloids in this contact zone, as well as intra-racial differences.
The supplementary material (pdfs) has plots for the significant components of the seven continental regions. Progress will now occur by extending population sampling to unexamined populations and by full-genome sequencing which will allow us to detect finer-level distinctions even in fairly homogeneous regions of the world such as Europe.
American Journal of Human Genetics doi:10.1016/j.ajhg.2009.04.015
Genome-wide Insights into the Patterns and Determinants of Fine-Scale Population Structure in Humans
Shameek Biswas et al.
Abstract
Studying genomic patterns of human population structure provides important insights into human evolutionary history and the relationship among populations, and it has significant practical implications for disease-gene mapping. Here we describe a principal component (PC)-based approach to studying intracontinental population structure in humans, identify the underlying markers mediating the observed patterns of fine-scale population structure, and infer the predominating evolutionary forces shaping local population structure. We applied this methodology to a data set of 650K SNPs genotyped in 944 unrelated individuals from 52 populations and demonstrate that, although typical PC analyses focus on the top axes of variation, substantial information about population structure is contained in lower-ranked PCs. We identified 18 significant PCs, some of which distinguish individual populations. In addition to visually representing sample clusters in PC biplots, we estimated the set of all SNPs significantly correlated with each of the most informative axes of variation. These polymorphisms, unlike ancestry-informative markers (AIMs), constitute a much larger set of loci that drive genomic signatures of population structure. The genome-wide distribution of these significantly correlated markers can largely be accounted for by the stochastic effects of genetic drift, although significant clustering does occur in genomic regions that have been previously implicated as targets of recent adaptive evolution.
Link
May 14, 2009
Admixture in Mexican Mestizos
Gene Expression points me towards a new open access paper in PNAS about genetic diversity in Mexican Mestizos:
The variation in individual admixture appears only somewhat stronger than in the Uyghur, which may be explained either by the smaller number of markers used here, making the assessment of admixture "noisier", or alternatively might be the result of the fact that admixture in Mexican Mestizos happened more recently, and immigration into the Americas from Europe continued hence, hence the homogenization of the population is still ongoing.
 Table S1 from the Supplementary material (pdf) shows the exact admixture proportions in the studied Mestizo populations and the HapMap populations.
Table S1 from the Supplementary material (pdf) shows the exact admixture proportions in the studied Mestizo populations and the HapMap populations.
Related:
PNAS doi:10.1073/pnas.0903045106
Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico
Irma Silva-Zolezzi et al.
Abstract
Mexico is developing the basis for genomic medicine to improve healthcare of its population. The extensive study of genetic diversity and linkage disequilibrium structure of different populations has made it possible to develop tagging and imputation strategies to comprehensively analyze common genetic variation in association studies of complex diseases. We assessed the benefit of a Mexican haplotype map to improve identification of genes related to common diseases in the Mexican population. We evaluated genetic diversity, linkage disequilibrium patterns, and extent of haplotype sharing using genomewide data from Mexican Mestizos from regions with different histories of admixture and particular population dynamics. Ancestry was evaluated by including 1 Mexican Amerindian group and data from the HapMap. Our results provide evidence of genetic differences between Mexican subpopulations that should be considered in the design and analysis of association studies of complex diseases. In addition, these results support the notion that a haplotype map of the Mexican Mestizo population can reduce the number of tag SNPs required to characterize common genetic variation in this population. This is one of the first genomewide genotyping efforts of a recently admixed population in Latin America.
Link
We analyzed data from 300 nonrelated self-identified Mestizo individuals from 6 states located in geographically distant regions in Mexico: Sonora (SON) and Zacatecas (ZAC) in the north, Guanajuato (GUA) in the center, Guerrero (GUE) in the center– Pacific, Veracruz (VER) in the center–Gulf, and Yucatan (YUC) in the southeast. Considering that Zapotecos have been shown as a good ancestral population for predicting Amerindian (AMI) ancestry in Mexican Mestizos (16), we included 30 Zapotecos (ZAP) from the southwestern state of Oaxaca (Fig. 1). For comparative purposes, we included similar data sets from HapMap populations: northern Europeans (CEU), Africans (YRI), and East Asians (EA), including Chinese (CHB) and Japanese (JPT).As expected, Mestizo admixture is mainly between Caucasoids and Amerindians, with a very little Sub-Saharan African thrown in at the individual level. Moreover, as with many populations, such as the Uyghur, where admixture took place several generations ago, individual admixture levels are fairly uniform, with very few individuals deviating strongly towards either the Caucasoid or Amerindian end of the spectrum.
The variation in individual admixture appears only somewhat stronger than in the Uyghur, which may be explained either by the smaller number of markers used here, making the assessment of admixture "noisier", or alternatively might be the result of the fact that admixture in Mexican Mestizos happened more recently, and immigration into the Americas from Europe continued hence, hence the homogenization of the population is still ongoing.
 Table S1 from the Supplementary material (pdf) shows the exact admixture proportions in the studied Mestizo populations and the HapMap populations.
Table S1 from the Supplementary material (pdf) shows the exact admixture proportions in the studied Mestizo populations and the HapMap populations.Related:
PNAS doi:10.1073/pnas.0903045106
Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico
Irma Silva-Zolezzi et al.
Abstract
Mexico is developing the basis for genomic medicine to improve healthcare of its population. The extensive study of genetic diversity and linkage disequilibrium structure of different populations has made it possible to develop tagging and imputation strategies to comprehensively analyze common genetic variation in association studies of complex diseases. We assessed the benefit of a Mexican haplotype map to improve identification of genes related to common diseases in the Mexican population. We evaluated genetic diversity, linkage disequilibrium patterns, and extent of haplotype sharing using genomewide data from Mexican Mestizos from regions with different histories of admixture and particular population dynamics. Ancestry was evaluated by including 1 Mexican Amerindian group and data from the HapMap. Our results provide evidence of genetic differences between Mexican subpopulations that should be considered in the design and analysis of association studies of complex diseases. In addition, these results support the notion that a haplotype map of the Mexican Mestizo population can reduce the number of tag SNPs required to characterize common genetic variation in this population. This is one of the first genomewide genotyping efforts of a recently admixed population in Latin America.
Link
35,000-year old female figurine from Hohle Fels Cave
 This is said to be the oldest currently known depiction of the human form. There is also a free video in the Nature site; see also a BBC story with more pics.
This is said to be the oldest currently known depiction of the human form. There is also a free video in the Nature site; see also a BBC story with more pics.From the paper:
The new figurine from Hohle Fels radically changes our view of the origins of Palaeolithic art. Before this discovery, animals and therianthropic imagery dominated the two dozen figurines from the Swabian Aurignacian. Female imagery was entirely unknown2, 15. With this discovery, the widespread notion that three-dimensional female depictions developed in the Gravettian can be rejected
Nature doi:10.1038/nature07995
A female figurine from the basal Aurignacian of Hohle Fels Cave in southwestern Germany
Nicholas J. Conard
Abstract
Despite well over 100 years of research and debate, the origins of art remain contentious1, 2, 3. In recent years, abstract depictions have been documented at southern African sites dating to approx75 kyr before present (bp)4, 5, and the earliest figurative art, which is often seen as an important proxy for advanced symbolic communication, has been documented in Europe as dating to between 30 and 40 kyr bp 2. Here I report the discovery of a female mammoth-ivory figurine in the basal Aurignacian deposit at Hohle Fels Cave in the Swabian Jura of southwestern Germany during excavations in 2008. This figurine was produced at least 35,000 calendar years ago, making it one of the oldest known examples of figurative art. This discovery predates the well-known Venuses from the Gravettian culture by at least 5,000 years and radically changes our views of the context and meaning of the earliest Palaeolithic art.
Link
May 13, 2009
Vitamin D and human depigmentation
This is a response to Robins et al. (2009)
Vitamin D and the evolution of human depigmentation
George Chaplin, Nina G. Jablonski
Abstract
In his recent commentary, Robins (2009) disputed the
role played by ultraviolet radiation (UVR), namely, the
vitamin-D-producing wavelengths of ultraviolet B (UVB),
in the evolution of human skin. He questioned the
theory that reduced levels of pigmentation in human
skin were selected to facilitate absorption of UVB. He
provided evidence to support his idea that people can
produce enough vitamin D in their skin, regardless of
pigmentation, if they are not pursuing a modern lifestyle.
He asserted that, within his framework, rickets
was the only selective force that could have influenced
the evolution of light pigmentation because other detrimental
effects of vitamin D deficiency are unproven. As
rickets is increased by industrialization, Robins concluded
that ‘‘. . . vitamin D status could not have constituted the
fitness differential between lightly pigmented and darkly
pigmented individuals at high latitudes that favored the
evolutionary selection of the former’’ (Robins, 2009).
In this article, we examine the current evidence for
what has been termed the ‘‘vitamin D theory,’’ and highlight
the importance of UVB penetration in the evolution
of human skin. We begin with an overview of the solar
processes involved in cutaneous vitamin D synthesis, followed
by a discussion of causal arguments and causation
in the context of the vitamin D theory, and conclude with
a review of physiological mechanisms and their evolutionary
significance.
Link
Robins’s (2008) [sic] opening quote from Huxley is an interesting 1870 antecedent of Popperian scientific philosophy. From a Kuhnian perspective (Kuhn, 1962), it does not apply very well to the paradigmatic role of vitamin D in the evolution of human skin pigmentation, because anomalies alone are insufficient to overthrow a paradigm without an alternative competing paradigm. As to the observation of a singular, linear correlation of skin pigmentation to UVB in the winter, the vitamin D hypothesis still holds explanatory scope and predictive success. Whatever be its specific actions, vitamin D is the only agent that can account for the observation that light skin is actively selected in areas where UVB is seasonal, absent, or more variable. In areas where UVB is strong and unwavering, dark skin is positively selected.Related:
- Skin color evolution in Europeans and Social skin color vs Disease in Puerto Ricans
- The Evolution of Human Skin and Skin Color
Vitamin D and the evolution of human depigmentation
George Chaplin, Nina G. Jablonski
Abstract
In his recent commentary, Robins (2009) disputed the
role played by ultraviolet radiation (UVR), namely, the
vitamin-D-producing wavelengths of ultraviolet B (UVB),
in the evolution of human skin. He questioned the
theory that reduced levels of pigmentation in human
skin were selected to facilitate absorption of UVB. He
provided evidence to support his idea that people can
produce enough vitamin D in their skin, regardless of
pigmentation, if they are not pursuing a modern lifestyle.
He asserted that, within his framework, rickets
was the only selective force that could have influenced
the evolution of light pigmentation because other detrimental
effects of vitamin D deficiency are unproven. As
rickets is increased by industrialization, Robins concluded
that ‘‘. . . vitamin D status could not have constituted the
fitness differential between lightly pigmented and darkly
pigmented individuals at high latitudes that favored the
evolutionary selection of the former’’ (Robins, 2009).
In this article, we examine the current evidence for
what has been termed the ‘‘vitamin D theory,’’ and highlight
the importance of UVB penetration in the evolution
of human skin. We begin with an overview of the solar
processes involved in cutaneous vitamin D synthesis, followed
by a discussion of causal arguments and causation
in the context of the vitamin D theory, and conclude with
a review of physiological mechanisms and their evolutionary
significance.
Link
May 12, 2009
Light-pigmented Caucasoids from prehistoric Siberia
 This sample was previously tested for Y-chromosome and mtDNA polymorphisms.
This sample was previously tested for Y-chromosome and mtDNA polymorphisms.The pigmentation-related loci tested can be seen in the labels of my post, which should lead you to some earlier studies on them.
Most individuals were found to be most similar to European than to East Asian or African individuals based on these loci, although some (2 from Andronovo) of them were more similar to East Asians or intermediate (1 from Tagar) between East Asians and Europeans.
Interestingly, 1 of the Andronovo Mongoloids (S07) was previously found to belong to Y chromosome haplogroup C(xC3), while the Caucasoid-Mongoloid individual from Tagar (S32) belonged to haplogroup R1a1.
It should be noted that the use of the term "European individual ancestry" does not mean that these individuals were from Europe, as no test to distinguish between European and Asian Caucasoids was performed, and we know from literary descriptions and occasional archaeological remains about the ancient presence of light-pigmented Caucasoids in Siberia.
From the paper:
The genotype for rs12913832 was obtained for 23 out of the 25 samples, and most had the G/G genotype (n=15), which indicates that at least 60% of ancient specimens were probably blue- or green-eyed individuals. The remaining samples had the A/G (n=5) or A/A (n=3) genotypes, which are predictive of brown eye color phenotype.
International Journal of Legal Medicine doi:10.1007/s00414-009-0348-5
Pigment phenotype and biogeographical ancestry from ancient skeletal remains: inferences from multiplexed autosomal SNP analysis
Caroline Bouakaze et al.
Abstract
In the present study, a multiplexed genotyping assay for ten single nucleotide polymorphisms (SNPs) located within six pigmentation candidate genes was developed on modern biological samples and applied to DNA retrieved from 25 archeological human remains from southern central Siberia dating from the Bronze and Iron Ages. SNP genotyping was successful for the majority of ancient samples and revealed that most probably had typical European pigment features, i.e., blue or green eye color, light hair color and skin type, and were likely of European individual ancestry. To our knowledge, this study reports for the first time the multiplexed typing of autosomal SNPs on aged and degraded DNA. By providing valuable information on pigment traits of an individual and allowing individual biogeographical ancestry estimation, autosomal SNP typing can improve ancient DNA studies and aid human identification in some forensic casework situations when used to complement conventional molecular markers.
Link
Y chromosome haplogroup E-M78 subtyping in Italians
The overall frequency of haplogroup E-M78 was 7.78% in the Piedmont and 11.44% in Siily, with E-V13 present at a frequency of 3.33% and 5.93% respectively. The Sicilian sample is the same as in di Gaetano et al.. The paper includes Y-STR minimum haplotype data in its supplementary material.
International Journal of Legal Medicine doi:10.1007/s00414-009-0350-y
Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application
S. Caratti et al.
Abstract
The continual discovery of new single-nucleotide polymorphisms (SNPs) has led to an increased resolution of the Y chromosome phylogeny. Some of these Y-SNPs have shown to be restricted to small geographical regions and therefore may prove useful in the forensic field as tools for the prediction of population of origin of unknown casework samples. Here, we describe a system for the molecular dissection of haplogroup E-M78 (E1b1b1a), consisting of multiplex polymerase chain reaction and minisequencing of M78 and nine population-informative Y-SNPs (M148, M224, V12, V13, V19, V22, V27, V32, V65) in a single reaction. Sensitivity and admixture studies demonstrated that the SNP protocol allows robust genotyping from as little as 50 pg of male DNA, even in the presence of 500-fold amounts of female DNA. In order to evaluate the suitability of E1b1b1a, subhaplogrouping for population-of-origin prediction, the distribution of E-M78 and its derived variants was determined in an Italian population sample (n = 326).
Link
International Journal of Legal Medicine doi:10.1007/s00414-009-0350-y
Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application
S. Caratti et al.
Abstract
The continual discovery of new single-nucleotide polymorphisms (SNPs) has led to an increased resolution of the Y chromosome phylogeny. Some of these Y-SNPs have shown to be restricted to small geographical regions and therefore may prove useful in the forensic field as tools for the prediction of population of origin of unknown casework samples. Here, we describe a system for the molecular dissection of haplogroup E-M78 (E1b1b1a), consisting of multiplex polymerase chain reaction and minisequencing of M78 and nine population-informative Y-SNPs (M148, M224, V12, V13, V19, V22, V27, V32, V65) in a single reaction. Sensitivity and admixture studies demonstrated that the SNP protocol allows robust genotyping from as little as 50 pg of male DNA, even in the presence of 500-fold amounts of female DNA. In order to evaluate the suitability of E1b1b1a, subhaplogrouping for population-of-origin prediction, the distribution of E-M78 and its derived variants was determined in an Italian population sample (n = 326).
Link
May 11, 2009
Injuries and Violence in Iberian Neolithic and Bronze Age
American Journal of Physical Anthropology doi:10.1002/ajpa.21089
Possible relationship of cranial traumatic injuries with violence in the south-east Iberian Peninsula from the Neolithic to the Bronze Age
S.A. Jiménez-Brobeil et al.
Abstract
The main aim of this study was to analyze the presence and distribution of cranial trauma, as possible evidence of violence, in remains from the Neolithic to Bronze Age from the SE Iberian Peninsula. The sample contains skulls, crania, and cranial vaults belonging to 410 prehistoric individuals. We also studied 267 crania from medieval and modern times for comparative purposes. All lesions in the prehistoric crania are healed and none of them can be attributed to a specific weapon. In all studied populations, injuries were more frequent in adults than in subadults and also in males than in females, denoting a sexual division in the risk of suffering accidents or intentional violence. According to the archeological record, the development of societies in the SE Iberian Peninsula during these periods must have entailed an increase in conflict. However, a high frequency of cranial traumatic injuries was observed in the Neolithic series, theoretically a less conflictive time, and the lowest frequency was in crania from the 3rd millennium B.C. (Copper Age), which is characterized by the archeologists as a period of increasing violence. The relatively large size and the high rate of injuries in Neolithic crania and the practice of cannibalism are strongly suggestive of episodes of interpersonal or intergroup conflict. The number and distribution of injuries in Bronze Age is consistent with the increase in violence at that time described by most archeologists.
Link
Possible relationship of cranial traumatic injuries with violence in the south-east Iberian Peninsula from the Neolithic to the Bronze Age
S.A. Jiménez-Brobeil et al.
Abstract
The main aim of this study was to analyze the presence and distribution of cranial trauma, as possible evidence of violence, in remains from the Neolithic to Bronze Age from the SE Iberian Peninsula. The sample contains skulls, crania, and cranial vaults belonging to 410 prehistoric individuals. We also studied 267 crania from medieval and modern times for comparative purposes. All lesions in the prehistoric crania are healed and none of them can be attributed to a specific weapon. In all studied populations, injuries were more frequent in adults than in subadults and also in males than in females, denoting a sexual division in the risk of suffering accidents or intentional violence. According to the archeological record, the development of societies in the SE Iberian Peninsula during these periods must have entailed an increase in conflict. However, a high frequency of cranial traumatic injuries was observed in the Neolithic series, theoretically a less conflictive time, and the lowest frequency was in crania from the 3rd millennium B.C. (Copper Age), which is characterized by the archeologists as a period of increasing violence. The relatively large size and the high rate of injuries in Neolithic crania and the practice of cannibalism are strongly suggestive of episodes of interpersonal or intergroup conflict. The number and distribution of injuries in Bronze Age is consistent with the increase in violence at that time described by most archeologists.
Link
May 10, 2009
ESHG 2009 abstracts
ESHG 2009 is in two weeks, and there are some very interesting abstracts, including a tantalizing new study on Y-chromosome haplogroup R1b1b2 (R-M269).
Phylogeography of human Y chromosome haplogroup R1b1b2 (R-M269) in Europe
Y chromosome haplogroup R1a is associated with prostate cancer risk among Macedonian males
The genetic position of Western Brittany (Finistère, France) in the Celtic Y chromosome landscape
Mitochondrial Genome Diversity in Tungusic-speaking Populations (Even and Evenki) and Resettlement of Arctic Siberia After the Last Glacial Maximum
X-chromosomal haplotypes in global human populations
Dissecting the genetic make-up of Central Eastern Sardinia using a high density set of sex and autosomal markers
Genetic differences between four European populations
European Lactase Persistence Allele is Associated With Increase in Body Mass Index
Phylogeography of human Y chromosome haplogroup R1b1b2 (R-M269) in Europe
F. Cruciani et al.Note that in the abstract below, the authors refer to Slavopaionians, not Macedonians.
The human Y chromosome haplogroup R1b1b2 (R-M269) displays an extremely wide geographic distribution within Europe, with a decreasing frequency cline from Iberia (frequencies up to 90%) towards the Balkans (usually less than 10%). Previous studies have proposed that the observed R1b1b2 frequency cline is due to a population expansion from an Iberian Ice-age refugium after the LGM (Malaspina et al. 1998; Semino et al. 2000).
In this study, we explored the phylogeography of the human Y chromosome haplogroup R1b1b2 by analyzing more than 2,000 males from Europe. The haplogroup-defining marker M269 (Cruciani et al. 2002), and two additional internal markers (U106 and U152, Sims et al 2007) which identify internal branches (R1b1b2g and R1b1b2h) were analyzed. The paragroup R1b1b2*(xR1b1b2g, R1b1b2h) and the haplogroups R1b1b2g and R1b1b2h showed quite different frequency distribution patterns within Europe, with frequency peaks in the Iberian Peninsula, northern Europe and northern Italy/France, respectively. The overall frequency pattern of R1b1b2 haplogroup is suggestive of multiple events of migration and expansion within Europe rather than a single and uniform spread of people from an Iberian Ice-age refugium.
References:
Malaspina et al. (1998) Am J Hum Genet 63:847-860
Semino et al. (2000) Science 290:1155-1159
Cruciani et al. (2002) Am J Hum Genet 70:1197-1214
Sims et al. (2007) Hum Mutat 28:97
Y chromosome haplogroup R1a is associated with prostate cancer risk among Macedonian males
D. Plaseska-Karanfilska et al.
Prostate cancer (PC) is one of the most common male-specific cancers. Its incidence varies considerably between populations. Recent surveys suggest that PC is influenced by both genetic and environmental factors, although the etiology of the disease remains unknown in the majority of cases. Certain Y chromosomal lineages have been suggested to predispose individuals to prostate cancer in Japanese population, but no association has been found among Korean and Swedish patients. The aim of this study was to investigate the association between Y chromosomal haplogroups and predisposition to prostate cancer in Macedonian men. We studied 84 PC patients and 126 males from the general population of Macedonian ethnic origin. A total of 28 markers have been studied by multiplex PCR and SNaPshot analysis. Nineteen different Y haplogroups were determined; the most frequent being I1b-P37b, E3b1-M78, R1a-SRY 1532, R1b-P25 and J2b1a-M241. The frequency of R1a was significantly higher in PC patients (20.2%) in comparison with the controls (9.5%) [p=0.027; OR=2.41 (1.09-5.36)]. When stratified according to age, even stronger association was observed between haplogroup R1a and prostate cancer in patients of >65 years of age [p=0.004; OR=3.24 (1.41-7.46)]. Our results suggest that Y chromosome haplogroup R1a is associated with an increased prostate cancer risk in Macedonian men.
The genetic position of Western Brittany (Finistère, France) in the Celtic Y chromosome landscape
K. Rouault et al.
Brittany, a large peninsula located at the western part of France, is of particular interest because of its historical settlement and its relative geographic and cultural isolation. Brittany was invaded by waves of migration from Britain and Ireland between the 4th and 7th centuries and, therefore, belongs to the Brythonic branch of the Insular Celtic language. We have focused our study on the department of Finistère, the most western territorial unit of Brittany, and its administrative and historical areas. To explore the diversity of the Y-chromosome, we analyzed a total of 348 unrelated males using a combination of 23 biallelic markers and 12 microsatellite loci. The molecular analysis revealed that 82.2% of the Y chromosomes fell into haplogroup R1b, placing Finistère within the Western European landscape. Interestingly, at a microgeographical level, differences were detected by the haplogroup R1a* being confined to the south of the department, while haplogroups E3b, F, G, J2, K and R1a1 were found in the north. Nevertheless, geographical distribution of haplogroups and haplotypes suggested territorial homogeneity inside Finistère. Most of the Y-chromosomal gene pool in Finistère is shared with European, especially British, populations, thus corroborating the historical reports of ancient migrations to Brittany. Finally, the results are consistent with those obtained from classic genetic markers and support the Celtic paternal heritage of the Finistère population.
Mitochondrial Genome Diversity in Tungusic-speaking Populations (Even and Evenki) and Resettlement of Arctic Siberia After the Last Glacial Maximum
I. O. Mazunin et al.
The present study includes the Even/Evenki, hunters and reindeer-breeders, sampled from a few localities scattered across their vast geographic range encompassing low Yana-Indigirka-Kolyma in the west and the Sea of Okhotsk coast in the east. The mtDNA data show a very close affinity of the Even/Evenki with the Yukaghir, typical reindeer hunters, dominating in extreme northeastern Siberia until the middle of 18th century but now being on the brink of extinction. We found that the majority of mtDNA diversity in the Tungusic-speaking populations was accounted for by Siberian-East Eurasian lineages C2, C3, D2, D3, D4-D9 and G1. The similarity in the haplogroup C and D mtDNA intrinsic variation between the Even and Yukaghir populations is pronounced and indicates that the Even/Evenki harbor an essential portion of the ancestral Yukaghir pool. The phylogeography of the D4-D9 point to an early Neolithic phase expansion initiated northward to the northern and eastern perimeters of former Beringia. Concerning unique D2* lineage (Volodko et al. 2008), the network analysis encompassing four complete sequences, three of the Yukaghir from the low Indigirka-Kolyma region and one of the Evenk from the upper reaches of the Aldan River would suggest that the founding haplotype (1935-8683-14905) for D2* originated within western part of former Beringia. In the meanwhile, the core of the Even/Evenki mtDNA pool residing in the midst of the Yukaghir ancient territory would represent a recent amalgamation of the remnants of the Yukaghir and northern Tungusic-speakers (Even/Evenki) originated in the mid-Amur region.
X-chromosomal haplotypes in global human populations
V. A. Stepanov, I. Y. Khitrinskaya;
To reconstruct the origin and evolution of X-chromosomal lineages in global human populations we investigated the genetic diversity in 23 population samples (about 1500 individuals totally) using SNP markers in a single linkage disequilibrium region of ZFX gene. About sixty haplotypes belonging to 3 phylogenetic branches (A, B, and F) originated from the single African root were found in the total sample. Branch A includes mostly African haplotypes, whereas four major haplotypes belonging to different sub-branches of B (haplotype E8) and F (haplotypes H4, I3 and I11) were present in Eurasia. Major haplotype of the older branch B (E8) is almost evenly distributed among Eurasian populations. Haplotypes of the younger phylogenetic branches demonstrates clinal distribution with the sharp frequency changes from East to West. Haplotype H4 is presumably “Eastern-Eurasian”. It reaches the highest frequency in Eastern and South-Eastern Asians. Haplotypes I3 and I11 in the contrary show the clear frequency gradient from West to East with the highest frequency in Europeans, moderate frequency in Central Asia, and the minimal frequency in North-East and South-East Asia. The total level of genetic differentiation of global human populations estimated by the analysis of molecular variance of X-chromosomal haplotypes (Fst = 9.1%) is quite high and roughly corresponds to those measured for most other types of genetic markers except Y-chromosomal haplogroups which are characterized by the much higher level of between-population differences.
Dissecting the genetic make-up of Central Eastern Sardinia using a high density set of sex and autosomal markers
L. M. Pardo
Genetic isolates are valuable for identifying genetic variations underlying complex traits. However, prior knowledge of the genetic structure of the isolate is fundamental for carrying-out genome-wide association studies (GWAS) in these populations. The Sardinian population is currently the target of GWAS because of its ancient origin and long-standing isolation. To perform GWAS in Sardinia, we aim to characterize a subpopulation from the archaic area of Central-Eastern Sardinia at the genomic level. We used sex-specific markers (Y-chromosome and mtDNA) to assess the heterogeneity of the founder lineages and the divergence from other populations. In addition, we used a dense set of autosomal markers (SNP 5.0 array, Affymetrix) to investigate genome-wide Linkage Disequilibrium, to construct a Copy Number Variation map and to estimate pair-wise kinship and inbreeding.We first determined Y-chromosome lineages in 256 unrelated Sardinians using biallelic and microsatellite markers. Our analysis showed that the frequency of the major Y haplogroups clearly sets this population apart from other European haplogroups. The analysis of microsatellite markers revealed a high degree of gene diversity. Pairwise kinship and inbreeding were estimated in 113 subjects using 77709 autosomal SNP markers. We found that 16% of the subject pairs shared identical-by descent alleles more often than expected by chance. Furthermore, 60% of the subjects had low inbreeding coefficient values. Our preliminary results confirm that Sardinia is genetically different from other populations, as shown by Y-chromosome markers. The kinship and inbreeding estimates indicate some degree of relatedness among Sardinians, as expected for an isolated population.
Genetic differences between four European populations
Genomic runs of homozygosity: population history and disease
V. Moskvina et al.
Population stratification can distort the results of genome-wide association studies (GWAS). One approach to deal with this inflation of the statistic is to estimate the inflation factor and adjust the detection statistic accordingly. However, the evolutionally forces work with different strength in some regions of the human genome, e.g. around the lactase gene (LCT) and the HLA region, making such an adjustment inappropriate.
We examined the population differences in four European populations (Scotland, Ireland, Sweden and Bulgaria) using data from GWAS performed with the Affymetrix 6.0 array at the Broad Institute. We show that there are >20,000 SNPs which are highly (p less than 10-6) significantly stratified between the four populations, after genome wide Bonferroni correction for multiple testing. We then examined the top 20 stratified regions to see what genes might have caused the top differences, using a highly conservative cut-off of p less than 10-40. Some of the loci span genes reported before: hair colour and pigmentation (HERC2, EXOC2), the LCT gene, genes involved in NAD metabolism, and genes involved in immunity (HLA and the Toll-like receptor genes TLR10, TLR 1, TLR 6). Among the top hits were several genes which have not yet been reported as stratified within European populations, indicating that they might also provide a selective advantage. Some involve other immunity genes (CD99, ILT6), but others show no obvious effect on positive selection: several zinc fingers, and most intriguingly, FOXP2, implicated in speech development. Future GWAS should take into consideration any positive associations with these genes.
R. McQuillan
Runs of homozygosity (ROH), resulting from the inheritance from both parents of identical haplotypes, are abundant in the human genome. ROH length is determined partly by the number of generations since the common ancestor: offspring of cousin matings have long ROH, while the numerous shorter ROH reflect shared ancestry tens and hundreds of generations ago. In studies of European populations we show that Froh, a multipoint estimate of individual autozygosity derived from genomic ROH, distinguishes clearly between subpopulations classified in terms of demographic history and correlates strongly with pedigree-derived inbreeding coefficients. In a global population dataset, analysis of ROH allows categorisation of individuals into four major groups, inferred to have (a) parental relatedness in the last 150 years (many south and west Asians), (b) shared parental ancestry arising hundreds to thousands of years ago through population isolation and restricted effective population size (Ne), but little recent inbreeding (Oceanians, African hunter-gatherers, some European and south Asian isolates), (c) both ancient and recent parental relatedness (Native Americans), and (d) only the background level of shared ancestry relating to continental Ne (east Asians, urban Europeans; African agriculturalists). Long runs of homozygosity are therefore a widespread and underappreciated characteristic of our genomes which record past consanguinity and population isolation and provide a unique record of individual demographic history. Individual ROH measures also allow quantification of the disease risk arising from polygenic recessive effects. We present preliminary data from a survey of the effects of ROH on quantitative disease-related traits and disease risk.
European Lactase Persistence Allele is Associated With Increase in Body Mass Index
J. A. Kettunen et al.
The global prevalence of obesity, usually indexed by body mass index (BMI) cut-offs, has increased significantly in the recent decades, mainly due to positive energy balance. However, the impact of a selection for specific genes cannot be excluded. Here we have tested the association between BMI and one of the best known genetic variants showing strong selective pressure: the functional variant in the cis-regulatory element of the lactase gene. We tested this variant since it is presumed to provide nutritional advantage in specific physical and cultural environments. We found that the variant responsible for lactase persistence among Europeans was also associated with higher BMI in a Nordic population sample (p = 1.3*10-5) of 15 209 individuals, the size of the effect being close to that of FTO. We tested the effect of population stratification and concluded that the association was not due to population substructure.
Subscribe to:
Posts (Atom)