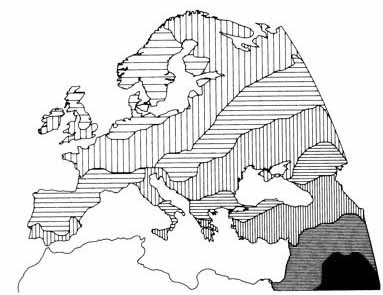
This map was drawn before carbon dating had been invented. We now know much more about both the anthropology and archaeology of Europe. But, the main thrust of Coon's prehistorical narrative can be summarizes as arrows on a map, or, prehistory as a series of invasions. The closing paragraph from the Neolithic Invasions chapter sums up this view admirably:
Today, carbon dating has pushed the arrival of the Neolithic to Europe into the 7th millennium BC, but, disregarding that detail, we can see that
farmers, seafarers and pastoralists from the south and east.
He did think that the Upper Paleolithic population had not disappeared completely, but the name he often used to describe them was
survivors, which denoted quite clearly their limited contribution to the present-day population.
Acculturation & Demic Diffusion
After WWII, the arrows on a map paradigm was no longer in fashion. The transition from the old to the new prehistory did not happen overnight, but two new intellectual fashions gained ground: acculturation and demic diffusion.
The proponents of acculturation were motivated by a reaction to the pots and skulls paradigm. To the idea that the spread of a new pottery type, or a new type of skull morphology indicated the spread of a people across the map, they countered that (i) pottery could be exchanged, copied, and traded without the movement of people, and (ii) that conclusions based on typological old-style anthropology were unsupportable, and the limitless malleability of the human skull was affirmed.
In some respects, the acculturation hypothesis represented a valid response to the excesses of the pots and skulls tradition. But, they went a bit too far in presenting a picture of complete stasis, in which European people, seemingly fixed to the ground, participated only in "networks of exchange", only ideas and goods flowed, and all differences in physical type across long time spans were ascribed invariably to responses (genetic or plastic) to new technologies, but almost never to the introduction of a new population element.
Demic diffusion is not as extreme as the pure acculturation hypothesis, but it replaces the model of invasions and migrations represented by arrows with a purposeless random walk. Demic diffusion has been argued on both archaeological and genetic grounds.
When Cavalli-Sforza and colleagues collected genetic data on modern Europeans, and subjected them to principal components' analysis made possible by modern computers, they discovered that
the first principal component of genetic variation was oriented on a southeast-northwest axis.
At roughly the same time, the widespread dating of Neolithic sites across Europe proved that there was a fairly regular advent of farming, with the earlier sites found in Greece, and the latest ones in the Atlantic fringe and northern Europe.
Demic diffusion was summoned to explain these phenomena. Neolithic farmers, the story goes, did not particularly want to colonize Europe. Europe was colonized as a side-effect of a random process in which farmers moved away from their parent's home, while their population numbers grew due to the increased productivity of the farming economy.
The process was not seen as one of population replacement, however. Rather, it was seen as a slow movement of a wave of advance, in which farmers mixed with hunter-gatherers, and some of them moved on to populate new lands beyond the farmer-hunter frontier. The model predicted that the technology would spread without large-scale population replacement, as the hunters' genes would make a substantial contribution to farmers' gene pools at the furthest end of their expansion.
The Paleolithic Europeans make a comeback
Whereas Cavalli-Sforza and his colleagues had looked at dozens of polymorphisms, their synthetic PC maps of Europe didn't come with dates or easy explanations. The observed clines in Europe may have been due to Paleolithic, Neolithic, or even recent historical events. While they were consistent with the Neolithic demic diffusion hypothesis, the possibility existed that they may have been formed either earlier, or later than the Neolithic.
The new approach by Sykes, Richards, and their colleagues, looked at just mtDNA, but due to its being inherited from mother to daughter without recombination, they could (i) estimate the age of the common ancestors of the "European mothers", (ii) study the patterns of geographical distribution of their descendants to infer when and where they may have lived. Hence, the various stories about Katrine, Ulrike, Helena, etc. in Sykes's book.
The conclusions of the new methodology were clear (at least to the authors' satisfaction):
This robustness to differing criteria for the exclusion of back-migration and recurrent mutation suggests that the Neolithic contribution to the extant mtDNA pool is probably on the order of 10%–20% overall. Our regional analyses support this, with values of 20% for southeastern, central, northwestern, and northeastern Europe. The principal clusters involved seem to have been most of J, T1, and U3, with a possible H component. This would suggest that the early-Neolithic LBK expansions through central Europe did indeed include a substantial demic component, as has been proposed both by archaeologists and by geneticists (Ammerman and Cavalli-Sforza 1984; Sokal et al. Sokal et al., 1991 RR Sokal, NL Ogden and C Wilson, Genetic evidence for the spread of agriculture in Europe by demic diffusion, Nature 351 (1991), pp. 143–144.1991). Incoming lineages, at least on the maternal side, were nevertheless in the minority, in comparison with indigenous Mesolithic lineages whose bearers adopted the new way of life.
The picture of continuity since the Paleolithic was further supported in the much briefer article by Semino et al. (
pdf) on
The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perspective. This study, based mostly on the observation of rough congruences of the European map with some Y-chromosome markers set the stage for most Y-chromosome work in Europe for the next decade.
In today's terminology, this paper suggested that, like mtDNA, most European Y-chromosomes were Paleolithic in origin, and belonged in haplogroups R1b, R1a, and I which repopulated Europe from refugia in Iberia, the Ukraine, and the Balkans, after the last glaciation. To this set were added Neolithic immigrants from the Middle East bearing haplogroups J, G, and E1b1b, and Northern Asian immigrants from the east bearing haplogroup N1c.
Unfortunately, we do not have Y-chromosome data of Paleolithic age to determine the veracity of this scenario. Given present-day distributions, we can be fairly certain of a European origin (but when?) of haplogroup I, of a non-European origin of haplogroup E1b1b (via North Africa or the Middle East), and of N1c. A non-European origin of the entire haplogroups J and G in West Asia also seems quite probable.
The house of cards collapses
The beauty of science is that new data can always falsify cozy and plausible scientific theories. In the case of European prehistory, this occurred due to a combination of craniometric, archaeological, and mtDNA data.
Pinhasi and von Cramon-Taubadel (2009) examined skulls from the early Central European Neolithic (Linearbandkeramik) and found them to be closer to Neolithic skulls from Balkans and West Asia, rather than the per-farming Mesolithic populations.
Our results demonstrate that the craniometric data fit a model of continuous dispersal of people (and their genes) from Southwest Asia to Europe significantly better than a null model of cultural diffusion.
The authors correctly identified their data as rejecting cultural diffusion, but their conclusion that they supported demic diffusion was not warranted as there was really no evidence that Neolithic groups were "transformed" by gradual slow admixture with hunter-gatherers in their march into Europe. Their data could just as easily be explained by plain migration.
Archaeologists also made a strong case for a rapid diffusion of the Neolithic in the Mediterranean. Neolithic settlements appeared suddenly, fully-formed, occupied regions abandoned by Mesolithic peoples, and spread not slowly, in a wave of advance, but rapidly, as a full-fledged colonization:
Thus it appears that none of the earlier models for Neolithic emergence in the Mediterranean accurately or adequately frame the transition. Clearly there was a movement of people westward out of the Near East all of the way to the Atlantic shores of the Iberian Peninsula. But this demic expansion did not follow the slow and steady, all encompassing pace of expansion predicted by the wave and advance model. Instead the rate of dispersal varied, with Neolithic colonists taking 2,000 years tomove from Cyprus to the Aegean, another 500 to reach Italy, and then only 500–600 years to travel the much greater distance from Italy to the Atlantic (52).
In a different study
Vanmontfort et al. studied the geographical distribution of farmers and hunter-gatherers during first contact in Central Europe. This contact did not involve either adoption of farming by hunter-gatherers (as in the acculturation hypothesis), or admixture with hunter-gatherers (as in the demic diffusion/wave of advance model).
Rather, agriculturalists and hunter-gatherers tended to avoid each other for 1,000 years after first contact!To conclude, the following model can be put forward. During the 6th Millennium cal BC, major parts of the loess region are exploited by a low density of hunter–gatherers. The LBK communities settle at arrival in locations fitting their preferred physical characteristics, but void of hunter–gatherer activity. Evidently, multiple processes and contact situations may have occurred simultaneously, but in general the arrival of the LBK did not attract hunter–gatherer hunting activity. Their presence rather restrained native activity to regions located farther away from the newly constructed settlements or triggered fundamental changes in the socio-economic organisation and activity of local hunter–gatherers. Evidence for the subsequent step in the transition dates to approximately one millennium later (Crombé and Vanmontfort, 2007; Vanmontfort, 2007).
The "Paleolithic" case won a short-lived victory when Haak et al tested mtDNA from early Central European farmers, discovering that they had a high frequency of haplogroup N1a which is rare in modern Europeans.
This finding was interpreted as evidence that the incoming Neolithic farmers were few in numbers and were absorbed with barely a trace by the surrounding Mesolithic populations who adopted agriculture. Acculturation seemed to have won the day! The case was, however, tentative, and hinged on the assumption that the Paleolithic Europeans -who had not been tested yet- would have a gene pool similar to that of modern Europeans.
When hunter-gatherer mtDNA was tested in both Scandinavia (by
Malmström et al) and Central/Eastern Europe (by
Bramanti et al.), it turned out that continuity from the Paleolithic was rejected.
Hunter-gatherers were dominated by mtDNA haplogroup U, and subgroups U4/U5 in particular. None of the other lineages postulated by Sykes et al. as being "Paleolithic" in origin were found in them. Moreover, there was substantial temporal overlap between hunter-gatherer and farmer cultures, but farmers seemed to lack mtDNA typical of hunter-gatherers and vice versa. Confirming the archaeological picture of the two groups avoiding each other, it now seemed that there was little genetic contact between the two, at least in the early age.
The Neolithic spread by newcomers; there was no acculturation of Mesolithic people; there was no slow process of admixture between farmer and hunter along a wave of advance.
The gap between contemporaneous farmer and hunter mtDNA gene pools was as large as that found between modern Europeans and native Australians! The whole controversy about the relative contributions of the Neolithic and Paleolithic in the modern European gene pool was found to be beside the point. The modern European gene pool did not seem to be particularly similar to either Paleolithic hunter or Neolithic farmer: it possessed any haplogroups completely absent in pre-Neolithic Europe. And, it did not have a high frequency of the N1a "signature" haplogroup of the Neolithic. Selection, migration, or a combination of both had reshaped the European gene pool from the Neolithic onwards.
Where things stand
We have come full circle. Once again, Paleolithic Europeans assume the status of survivors, as their typical lineages are observed in a small minority of modern Europeans. The evidence for widespread acculturation of European hunter-gatherers or their significant genetic contribution to incoming farmers along a wave of advance is just not there. Hunters and farmers possessed distinctive gene pools, and farmers expanded with barely a trace of absorption of hunter gene pools.
Clearly many details remain to be filled out. What does seem certain, however, is that dramatic events took place starting at the Neolithic, and that modern Europeans trace their ancestry principally to Neolithic and post-Neolithic migrants, and not to the post-glacial foragers who inhabited the continent.

 From the paper:
From the paper:



