One of the lessons from the following paper is that microsatellite diversity does not always point to the correct direction of migration. If we knew nothing about Brazilians and Portuguese, we might be inclined to attribute the lower Portuguese genetic diversity to migration from Brazil to Portugal, even though the opposite is the case.
Genetica Volume 126, Numbers 1-2 Pages: 251 - 260
Y Chromosome Diversity in Brazilians: Switching Perspectives from Slow to Fast Evolving Markers
Denise R. Carvalho-Silva et al.
We have previously shown that the Y chromosomes of ‘white’ Brazilians have their immediate geographical origin in Europe, with low frequency of sub-Saharan African chromosomes and virtual absence of Amerindian contribution. The typing of slow evolving polymorphisms on the Y chromosome also revealed no differences between Brazilians and Portuguese, the bulk of European immigrants to Brazil, and even among Brazilians from distinct regions of Brazil, the latter being in sharp contrast with mtDNA data. In order to test if the lack of differentiation is a sex-biased and not a marker-biased phenomenon, we decided to study faster evolving Y chromosome markers in samples from Brazil and Portugal previously studied. The population structure revealed by this work confirmed that there were indeed no significant differences between Brazil and Portugal and no population differentiation within the four geographical regions of Brazil, suggesting that this phenomenon is unrelated to the nature of the markers typed. Nevertheless the fast evolving markers did uncover a higher within population diversity in Brazil than Portugal, which could be explained by the input of diverse European Y chromosomes carried by several migration waves to Brazil. Our present data highlight the significance of typing and combining Y markers that evolve according to distinct mutational paces to usefully assess the levels of diversity in a given population, and can be applied in the study of populations derived from distinct geographical origins such as the Brazilians.
Link

February 28, 2006
No significant crypto-Jewish ancestry in Spanish Americans
A new paper debunks the claim that Spanish-Americans are significantly crypto-Jewish, i.e., descended from Spanish Jews who hid their Jewish status to blend in, and eventually even forgot their origin.
Ann Hum Biol. 2006 Jan-Feb;33(1):100-11.
Toward resolution of the debate regarding purported crypto-Jews in a Spanish-American population: Evidence from the Y chromosome.
Sutton WK, Knight A, Underhill PA, Neulander JS, Disotell TR, Mountain JL.
Background: The ethnic heritage of northernmost New Spain, including present-day northern New Mexico and southernmost Colorado, USA, is intensely debated. Local Spanish-American folkways and anecdotal narratives led to claims that the region was colonized primarily by secret- or crypto-Jews. Despite ethnographic criticisms, the notion of substantial crypto-Jewish ancestry among Spanish-Americans persists. Aim: We tested the null hypothesis that Spanish-Americans of northern New Mexico carry essentially the same profile of paternally inherited DNA variation as the peoples of Iberia, and the relevant alternative hypothesis that the sampled Spanish-Americans possess inherited DNA variation that reflects Jewish ancestry significantly greater than that in present-day Iberia. Subjects and Methods: We report frequencies of 19 Y-chromosome unique event polymorphism (UEP) biallelic markers for 139 men from across northern New Mexico and southern Colorado, USA, who self-identify as 'Spanish-American'. We used three different statistical tests of differentiation to compare frequencies of major UEP-defined clades or haplogroups with published data for Iberians, Jews, and other Mediterranean populations. We also report frequencies of derived UEP markers within each major haplogroup, compared with published data for relevant populations. Results: All tests of differentiation showed that, for frequencies of the major UEP-defined clades, Spanish-Americans and Iberians are statistically indistinguishable. All other pairwise comparisons, including between Spanish-Americans and Jews, and Iberians and Jews, revealed highly significant differences in UEP frequencies. Conclusion: Our results indicate that paternal genetic inheritance of Spanish-Americans is indistinguishable from that of Iberians and refute the popular and widely publicized scenario of significant crypto-Jewish ancestry of the Spanish-American population.
Link
Ann Hum Biol. 2006 Jan-Feb;33(1):100-11.
Toward resolution of the debate regarding purported crypto-Jews in a Spanish-American population: Evidence from the Y chromosome.
Sutton WK, Knight A, Underhill PA, Neulander JS, Disotell TR, Mountain JL.
Background: The ethnic heritage of northernmost New Spain, including present-day northern New Mexico and southernmost Colorado, USA, is intensely debated. Local Spanish-American folkways and anecdotal narratives led to claims that the region was colonized primarily by secret- or crypto-Jews. Despite ethnographic criticisms, the notion of substantial crypto-Jewish ancestry among Spanish-Americans persists. Aim: We tested the null hypothesis that Spanish-Americans of northern New Mexico carry essentially the same profile of paternally inherited DNA variation as the peoples of Iberia, and the relevant alternative hypothesis that the sampled Spanish-Americans possess inherited DNA variation that reflects Jewish ancestry significantly greater than that in present-day Iberia. Subjects and Methods: We report frequencies of 19 Y-chromosome unique event polymorphism (UEP) biallelic markers for 139 men from across northern New Mexico and southern Colorado, USA, who self-identify as 'Spanish-American'. We used three different statistical tests of differentiation to compare frequencies of major UEP-defined clades or haplogroups with published data for Iberians, Jews, and other Mediterranean populations. We also report frequencies of derived UEP markers within each major haplogroup, compared with published data for relevant populations. Results: All tests of differentiation showed that, for frequencies of the major UEP-defined clades, Spanish-Americans and Iberians are statistically indistinguishable. All other pairwise comparisons, including between Spanish-Americans and Jews, and Iberians and Jews, revealed highly significant differences in UEP frequencies. Conclusion: Our results indicate that paternal genetic inheritance of Spanish-Americans is indistinguishable from that of Iberians and refute the popular and widely publicized scenario of significant crypto-Jewish ancestry of the Spanish-American population.
Link
February 27, 2006
A textbook debate over Hinduism
This LA Times story is about changes in California schoolbooks requested by Hindu organizations. The interesting paragraph:
The latest research actually reinforces the Aryan Invasion Theory. According to that theory, the caste system is not a simple "division of labor" as the revisionists would suggest, but rather a social structure imposed by an intrusive group. A prediction of that theory is that the upper caste in the Hindu system would carry a greater genetic legacy of non-South Asian ancestry. This prediction is supported by current evidence.
The indigenist school must explain why Brahmins are more "West Eurasian" genetically, if they were just assigned this role in a grand within-India "division of labor". A more parsimonious explanation is that they are more "West Eurasian" genetically because, well, their ancestors came from West Eurasia.
Moreover, the "division of labor" theory could accommodate exogenous origins for some Indian castes, but it does not explain why the elite group also happens to coincide with the exogenous group, namely the Brahmin caste. A more parsimonious explanationis that the elite group is also the exogenous group, because a group of outsiders took control of Indian society and placed themselves on top.
The most contentious issue involves the origins of Hinduism. The common historical view, included in all textbooks, is that Indo-Europeans from Central Asia, called Aryans, migrated to India and laid the faith's foundation. But Bajpai and the Hindu groups hotly dispute the idea of any Aryan migration, citing new DNA evidence for their view that Hinduism developed indigenously. They have asked that textbooks include both views.There is of course no new DNA evidence that Hinduism developed indigenously. The latest studies suggest that Indians are of largely indigenous origin, but that does not mean that their religion is. Hinduism is a blend of many elements, and the contribution of local elements in it should be acknowledged and celebrated, but the binding thread is the religion of the Vedic Indo-Aryans, Sanskrit, and the Brahmin caste.
The latest research actually reinforces the Aryan Invasion Theory. According to that theory, the caste system is not a simple "division of labor" as the revisionists would suggest, but rather a social structure imposed by an intrusive group. A prediction of that theory is that the upper caste in the Hindu system would carry a greater genetic legacy of non-South Asian ancestry. This prediction is supported by current evidence.
The indigenist school must explain why Brahmins are more "West Eurasian" genetically, if they were just assigned this role in a grand within-India "division of labor". A more parsimonious explanation is that they are more "West Eurasian" genetically because, well, their ancestors came from West Eurasia.
Moreover, the "division of labor" theory could accommodate exogenous origins for some Indian castes, but it does not explain why the elite group also happens to coincide with the exogenous group, namely the Brahmin caste. A more parsimonious explanationis that the elite group is also the exogenous group, because a group of outsiders took control of Indian society and placed themselves on top.
Cranial size and shape
S. P. Pickering (Correlation of brain and head measurements, and relation of brain shape and size to shape and size of the head, American Journal of Physical Anthropology, Volume 15, Issue 1, Date: October/December 1930, Pages: 1-52):
Why are brachycephalic heads more capacious? I don't think that anyone has discovered the exact reasons why this is the case, but here is my theory:
In general, human heads are longer than they are broad, and broader than they are high. Heads of course, like any other organ, are expensive things (developmentally), and this is particularly the case for humans where the head forms a substantially larger part of the body than in other mammals.
During human evolution, cranial capacity has increased. Cranial capacity is of course correlated with gross size: bigger people have a bigger cranial cavity, and a bigger brain.
However, things are not that simple. It can be easily proven that different shapes with the same surface area have a different capacity. In two dimensions, ellipses and ovals have a smaller capacity than round shapes with the same perimeter.
Thus, a broad head achieves a greater volume than a long one of equal surface. Since surface corresponds to bone mass, which corresponds to developmental cost, it is possible that a rounder head shape is more economical: it achieves the same volume with smaller cost.
The same is also true for the height of the skull. Since skulls tend to be broader than they are high, a skull that expands on the vertical plane will tend to approach a more spherical shape.
It is thus not surprising that according to Lahr and Wright's useful definition:
It is general knowledge that an increase in brain size runs parallel with an increase in head size and with a change from dolichocephaly to brachycephaly as we ascend the scale from the anthropoids to modern man. This appears primarily to be a volume phenomenon.A. Thomson (quoted in the above paper) notes that:
Given a cavity of oval or ellptical form with elastic walls, the more its contents are increased the greater will be the tendency to assume a spherical shape.Pickering studied cadavers and their weight and volume directly, rather than relying on estimations from cephalic measurements:
The specimens used in this work gave an average capacity for dolichocephalic skulls of 1402.8 cc.; for mesocephalic, 1474.8 and for brachycephalic, 1520.6 cc.Interestingly, according to K. Beals et al. (Brain size, cranial morphology, climate, and time machines. Current Anthropology, 25, 301-330. ) there is a +0.37 correlation between cranial capacity and the cephalic index, i.e., broader-headed, more brachycephalic populations also have bigger cranial capacities.
The same tendency was shown in the size of the brains. The average size of the brains in this series was for the dolichocephalic heads, 1135.4 cc.; for the mesocephalic, 1144.2 cc., and for the brachycephalic, 1180.7 cc.
The figures show a marked increase from the dolichocephalic to the brachycephalic type. Though the number of specimens of the dolichocephalic type is not sufficient to be conclusive proof, the fact that brachycephaly runs parallel with an increase in brain and head size is very evident.
Why are brachycephalic heads more capacious? I don't think that anyone has discovered the exact reasons why this is the case, but here is my theory:
In general, human heads are longer than they are broad, and broader than they are high. Heads of course, like any other organ, are expensive things (developmentally), and this is particularly the case for humans where the head forms a substantially larger part of the body than in other mammals.
During human evolution, cranial capacity has increased. Cranial capacity is of course correlated with gross size: bigger people have a bigger cranial cavity, and a bigger brain.
However, things are not that simple. It can be easily proven that different shapes with the same surface area have a different capacity. In two dimensions, ellipses and ovals have a smaller capacity than round shapes with the same perimeter.
Thus, a broad head achieves a greater volume than a long one of equal surface. Since surface corresponds to bone mass, which corresponds to developmental cost, it is possible that a rounder head shape is more economical: it achieves the same volume with smaller cost.
The same is also true for the height of the skull. Since skulls tend to be broader than they are high, a skull that expands on the vertical plane will tend to approach a more spherical shape.
It is thus not surprising that according to Lahr and Wright's useful definition:
...it is generally agreed that a modern skull should present a relatively small face tucked under the vault that is relatively short and high, a relatively vertical forehead, parietal enlargement, a relatively rounded occiput, a flexed cranial base, a canine fossa, an occipital protuberance in the occipital bone and mental eminence or chin. Most descriptions of modern H. sapiens would also include skeletal gracility as characterizing the group.or that according to K. L. Beals (Climate and the evolution of brachycephalization, American Journal of Physical Anthropology, Volume 62, Issue 4, Date: December 1983, Pages: 425-437):
During the Holocene, the [cranial] index increases under all climatic conditions.Hopefully, a new generation of scientists will overcome the knee-jerk reaction against studying cranial variation and cognitive function, perhaps by relying on cranial models built with 3D digitizers rather than the older methods based on lengths, arcs, and indices.
...
Turning to the Pleistocene, the mean hominid cranial index has increased 9 units.
February 25, 2006
Rare haplotypes in mtDNA
Hum Biol. 2005 Aug;77(4):443-56
Rare haplotypes in mtDNA: applications in the analysis of biosocial aspects of past human populations.
Izagirre N, Alzualde A, Alonso S, Paz L, Alonso A, de la Rua C.
We report on the use of rare mutations to tackle biosocial questions such as kinship and differential burial practices from past human populations. To do this, we have inferred nucleotide position 73 of HVS-II in individuals classified as belonging to haplogroup H from 76 human dental samples from the necropolis of Aldaieta (Basque Country, Spain, 6th-7th century) by means of PCR and restriction enzyme tests. The same analysis has been performed for 146 extant individuals from the northern Iberian peninsula. A combination of haplotype H and 73G in HVS-II, rare in extant populations (0.5-3%), has been found at a frequency of 20% in the ancient population of Aldaieta. These data can be explained in terms of the existence of different burial practices associated with a variety of factors, mainly social status and kinship. This hypothesis is also supported by archeological data. These results indicate that caution should be taken when making phylogenetic inferences from extinct populations, because an uncharacterized kinship can significantly bias allele frequencies.
Link
Rare haplotypes in mtDNA: applications in the analysis of biosocial aspects of past human populations.
Izagirre N, Alzualde A, Alonso S, Paz L, Alonso A, de la Rua C.
We report on the use of rare mutations to tackle biosocial questions such as kinship and differential burial practices from past human populations. To do this, we have inferred nucleotide position 73 of HVS-II in individuals classified as belonging to haplogroup H from 76 human dental samples from the necropolis of Aldaieta (Basque Country, Spain, 6th-7th century) by means of PCR and restriction enzyme tests. The same analysis has been performed for 146 extant individuals from the northern Iberian peninsula. A combination of haplotype H and 73G in HVS-II, rare in extant populations (0.5-3%), has been found at a frequency of 20% in the ancient population of Aldaieta. These data can be explained in terms of the existence of different burial practices associated with a variety of factors, mainly social status and kinship. This hypothesis is also supported by archeological data. These results indicate that caution should be taken when making phylogenetic inferences from extinct populations, because an uncharacterized kinship can significantly bias allele frequencies.
Link
February 23, 2006
Colonization of Europe by modern humans was more rapid than previously thought
Friom the paper:
These problems can be overcome by using new pre-treatment techniques that remove newer carbon contaminants, and by making use of stratified carbon samples taken from deep-sea sediments.
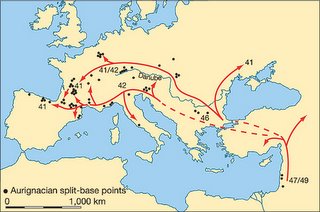
This is all very interesting, but it should be noted that the author's inferences are based on radiocarbon dating of Aurignacian remains. While it is generally believed that the Aurignacian was a creation of anatomically modern humans, there is no smoking gun link between the Aurignacian and modern human remains. In fact, it could well be argued that pushing the date of the Aurignacian further back in time may lend some support to the idea that Neandertals produced at least some of the Aurignacian assemblages.
Nature 439, 931-935 (23 February 2006) | doi:10.1038/nature04521
A new radiocarbon revolution and the dispersal of modern humans in Eurasia
Paul Mellars
Abstract
Radiocarbon dating has been fundamental to the study of human cultural and biological development over the past 50,000 yr. Two recent developments in the methodology of radiocarbon dating show that the speed of colonization of Europe by modern human populations was more rapid than previously believed, and that their period of coexistence with the preceding Neanderthal was shorter.
Link
The application of radiocarbon dating to these crucial early phases in modern human development has, however, been critically dependent on two potential sources of error in the accuracy of radiocarbon age estimates. The first is the impact of even miniscule quantities of contamination by more recent, intrusive carbon into the dated samples (Fig. 1a). This can be illustrated by the fact that contamination by only 1% of modern carbon in a sample actually 40,000 yr in age would reduce the measured age of the sample by over 7,000 yr... The second is the long-established recognition that the original proportion of 14C to 12C in the Earth's atmosphere has not remained constant over the past 50,000 yr, but has diverged sharply from present-day values, principally due to past variations in the intensity of the Earth's magnetic field and the shorter-term effects of sunspots on the amount of cosmic radiation reaching the upper atmosphere.
These problems can be overcome by using new pre-treatment techniques that remove newer carbon contaminants, and by making use of stratified carbon samples taken from deep-sea sediments.
The rapid spread of the early modern human populations was probably facilitated by a major improvement in climatic conditions in Europe between about 43,000–41,000 yr (calibrated) bp (the period of the Hengelo interstadial), which would inevitably have made a process of population expansion from southeast to northwest across Europe easier to achieve

This is all very interesting, but it should be noted that the author's inferences are based on radiocarbon dating of Aurignacian remains. While it is generally believed that the Aurignacian was a creation of anatomically modern humans, there is no smoking gun link between the Aurignacian and modern human remains. In fact, it could well be argued that pushing the date of the Aurignacian further back in time may lend some support to the idea that Neandertals produced at least some of the Aurignacian assemblages.
Nature 439, 931-935 (23 February 2006) | doi:10.1038/nature04521
A new radiocarbon revolution and the dispersal of modern humans in Eurasia
Paul Mellars
Abstract
Radiocarbon dating has been fundamental to the study of human cultural and biological development over the past 50,000 yr. Two recent developments in the methodology of radiocarbon dating show that the speed of colonization of Europe by modern human populations was more rapid than previously believed, and that their period of coexistence with the preceding Neanderthal was shorter.
Link
Iberian Origins of New World horses
J Hered. 2006 Feb 17; [Epub ahead of print]
Iberian Origins of New World Horse Breeds.
Luis C, Bastos-Silveira C, Cothran EG, Oom MD.
Fossil records, archaeological proofs, and historical documents report that horses persisted continuously in the Iberian Peninsula since the Pleistocene and were taken to the American continent (New World) in the 15th century. To investigate the variation within the mitochondrial DNA (mtDNA) control region of Iberian and New World horse breeds, to analyze their relationships, and to test the historical origin of New World horses, a total of 153 samples, representing 30 Iberian and New World breeds, were analyzed by sequencing mtDNA control region fragments. Fifty-four haplotypes were found and assigned to seven haplogroups. Reduced levels of variation found for the Menorquina, Sorraia, and Sulphur Mustang breeds are consistent with experienced bottlenecks or limited number of founders. For all diversity indices, Iberian breeds showed higher diversity values than South American and North American breeds. Although, the results show that the Iberian and New World breeds stem from multiple origins, we present a set of genetic data revealing a high frequency of Iberian haplotypes in New World breeds, which is consistent with historical documentation.
Link
Iberian Origins of New World Horse Breeds.
Luis C, Bastos-Silveira C, Cothran EG, Oom MD.
Fossil records, archaeological proofs, and historical documents report that horses persisted continuously in the Iberian Peninsula since the Pleistocene and were taken to the American continent (New World) in the 15th century. To investigate the variation within the mitochondrial DNA (mtDNA) control region of Iberian and New World horse breeds, to analyze their relationships, and to test the historical origin of New World horses, a total of 153 samples, representing 30 Iberian and New World breeds, were analyzed by sequencing mtDNA control region fragments. Fifty-four haplotypes were found and assigned to seven haplogroups. Reduced levels of variation found for the Menorquina, Sorraia, and Sulphur Mustang breeds are consistent with experienced bottlenecks or limited number of founders. For all diversity indices, Iberian breeds showed higher diversity values than South American and North American breeds. Although, the results show that the Iberian and New World breeds stem from multiple origins, we present a set of genetic data revealing a high frequency of Iberian haplotypes in New World breeds, which is consistent with historical documentation.
Link
Surnames and Y chromosomes
An interesting surname study shows that the probability of a surname match decreases as the surname frequency increases. In other words, it's quite unlikely that men with a common surname, say "Smith" will share a patrilineal ancestor, but very likely if the surname is rare.
Moreover, surname studies have forensic implications. By studying the Y chromosome it is possible to limit search to a few promising surnames. Of course, catching rapists is a worthwhile cause, but the recent success of an adopted teen to track down his biological father with Y-DNA testing shows, there are more sinister uses of this technology.
Curr Biol. 2006 Feb 21;16(4):384-388.
Genetic Signatures of Coancestry within Surnames.
King TE, Ballereau SJ, Schurer KE, Jobling MA.
Surnames are cultural markers of shared ancestry within human populations. The Y chromosome, like many surnames, is paternally inherited, so men sharing surnames might be expected to share similar Y chromosomes as a signature of coancestry. Such a relationship could be used to connect branches of family trees , to validate population genetic studies based on isonymy , and to predict surname from crime-scene samples in forensics . However, the link may be weak or absent due to multiple independent founders for many names, adoptions, name changes and nonpaternities, and mutation of Y haplotypes. Here, rather than focusing on a single name , we take a general approach by seeking evidence for a link in a sample of 150 randomly ascertained pairs of males who each share a British surname. We show that sharing a surname significantly elevates the probability of sharing a Y-chromosomal haplotype and that this probability increases as surname frequency decreases. Within our sample, we estimate that up to 24% of pairs share recent ancestry and that a large surname-based forensic database might contribute to the intelligence-led investigation of up to approximately 70 rapes and murders per year in the UK. This approach would be applicable to any society that uses patrilineal surnames of reasonable time-depth.
Link
February 22, 2006
Non-flawed mtDNA datasets
Nasidze and Stoneking respond to criticism levelled against them (see Flawed mtDNA datasets). I can't take any position on who is right or wrong, but feel free to read both criticism and response and make up your own mind. This little tidbit is interesting:
Annals of Human Genetics (Online Early)
The Patient is Not Dead Yet: Premature Autopsy of a mtDNA Data Set
M. Stoneking and I. Nasidze
(no abstract)
Link
B&K base their analyses on a database of 30,000 HV1 sequences, which they claim is sufficient to identify "… idiosyncrasies of data sets that cannot be attributed to natural evolutionary processes." We disagree: 30,000 sequences may sound like a lot, but actually this represents only 0.0005% of the potential HV1 sequences in the human population. Moreover, the existing sequences are nowhere near a random sample, as many areas of the world are under-represented in the studies that have been done to date. Given that 99.9995% of the data is missing, and that large areas of the world are missing in the 0.0005% of the data that does exist, it is hardly surprising that new patterns of variation will be found in new populations.
Annals of Human Genetics (Online Early)
The Patient is Not Dead Yet: Premature Autopsy of a mtDNA Data Set
M. Stoneking and I. Nasidze
(no abstract)
Link
February 21, 2006
Expansion of East Asian populations
Genetics. 2006 Feb 19; [Epub ahead of print]
Male demography in East Asia: a north-south contrast in human population expansion times.
Y. Xue et al.
The human population has increased greatly in size in the last 100,000 years, but the initial stimuli to growth, the times when expansion started, and their variation between different parts of the world are poorly understood. We have investigated male demography in East Asia, applying a Bayesian full-likelihood analysis to data from 988 men representing 27 populations from China, Mongolia, Korea and Japan typed with 45 binary and 16 STR markers from the Y chromosome. According to our analysis, the northern populations examined all started to expand in number between 34 (18-68) and 22 (12-39) thousand years ago (KYA), before the Last Glacial Maximum at 21-18 KYA, while the southern populations all started to expand between 18 (6-47) and 12 (1-45) KYA, but then grew faster. We suggest that the northern populations expanded earlier because they could exploit the abundant megafauna of the 'Mammoth Steppe', while the southern populations could only increase in number when a warmer and more stable climate led to more plentiful plant resources such as tubers.
Link
Male demography in East Asia: a north-south contrast in human population expansion times.
Y. Xue et al.
The human population has increased greatly in size in the last 100,000 years, but the initial stimuli to growth, the times when expansion started, and their variation between different parts of the world are poorly understood. We have investigated male demography in East Asia, applying a Bayesian full-likelihood analysis to data from 988 men representing 27 populations from China, Mongolia, Korea and Japan typed with 45 binary and 16 STR markers from the Y chromosome. According to our analysis, the northern populations examined all started to expand in number between 34 (18-68) and 22 (12-39) thousand years ago (KYA), before the Last Glacial Maximum at 21-18 KYA, while the southern populations all started to expand between 18 (6-47) and 12 (1-45) KYA, but then grew faster. We suggest that the northern populations expanded earlier because they could exploit the abundant megafauna of the 'Mammoth Steppe', while the southern populations could only increase in number when a warmer and more stable climate led to more plentiful plant resources such as tubers.
Link
Ancient Greek surgery is older than we thought

Anagnostis Aggelarakis has a very interesting piece in Archaeology Magazine detailing his study of a skull from the Greek colony of Abdera. The woman had received a blow by a missile to the back of the head which fractured her skull, but a type of surgery that is first described in the Hippocratic corpus, dating from the 5th-4th centuries BC, was able to save her life. The woman was thus able to live for another twenty years.
Artful Surgery (excerpt):
But new evidence, on which the story of the wounded young woman at the head of this article is based, will rewrite our history of the development of ancient medical practice. The patient was among those sent north by Clazomenae, a Greek city in Ionia, to establish a colony at Abdera around 654 B.C. She was successfully treated--a difficult operation performed by a master surgeon saved her--and lived for another 20 years. Her remains, which were excavated at Abdera by Eudokia Skarlatidou of the Greek Archaeological Service and which I have had the privilege to study, provide incontrovertible evidence that two centuries before Hippocrates drew breath, surgical practices described in the treatise On Head Wounds were already in use.
On Head Wounds sets forth diagnostic procedures for identifying and treating a range of cranial injuries caused by different weapons. In most cases, a wound on the back of the head--"where the bone is thicker and oozing puss will take longer to reach the brain"--was less likely to be fatal than one in the front. But, as in the case of the woman from Abdera, "When a suture shows at the exposed bone area of the wound--of a wound anywhere on the head--the resistance of the bone to the traumatic impact is very weak should the weapon get wedged in the suture." So, according to the Hippocratic text, the case was a serious one. It was made more so because of the nature of the weapon, a missile from a sling, because, "Of those weapons that strike the head and wound close to the cranial bone and the cranium itself, that one that will fall from a highest level rather than from a trajectory parallel to the ground, and being at the same time the hardest, bluntest, and heaviest...will crack and compress the cranial bone."
For compressed head fractures, On Head Wounds recommends trepanation, removal of a disk of bone from the skull using a drill with a serrated circular bit. This would eliminate the danger of bone splinters and radiating fracture fissures. It would also permit the removal of bone fragments that had been crushed inward, allowing the brain to swell from the contusion without pressing against loose bone fragments with sharp edges that might puncture the dura mater. But there was one cranial area where a scraping approach was strongly recommended instead of trepanation: "It is necessary, if the wound is at the sutures and the weapon penetrated and lodged into the bone, to pay attention for recognizing the kind of injury sustained by the bone. Because...he who received the weapon at the sutures will suffer far greater impact at the cranial bone than the one who did not receive it at the sutures. And most of those require trepanation, but you must not trepan the sutures themselves...you are required to scrape the surface of the cranial bone with a rasp in depth and length, according to the position of the wound, and then cross-wise to be able to see the hidden breakages and crushes...because scraping exposes the harm well, even if those injuries...were not otherwise revealed."
Faced with a compressed fracture with radiating fissure fractures and fearing damage to the dura mater, the surgeon scraped the bone in length, width, and depth, removing fragments and eliminating the fissures through scraping and not trepanation. He then would have tended to any adjacent injured tissues.
While the reconstruction of the patient's treatment is in part conjecture, based on the Hippocratic text itself, the size and shape of the surgical intervention and use of the rasp rather than trepanation is certain from traces on the bone itself. So the surgical procedure matches perfectly what was recommended two centuries later in On Head Wounds for this type of injury in this location.
We do not know exactly how the Clazomeneans chose the colonists who sailed to Abdera. Were they an elite group, the less wealthy who were willing to risk the venture, or the politically and socially disfavored? We do know, however, that among them there was a masterful surgeon, Hippocrates' predecessor, who was among the earliest of the Ionian school of medical practitioners.
February 20, 2006
More on CASP12 evolution
Afarensis points me to another new paper that deals with evolution in the CASP12 gene. As I detailed in my previous post, the inactive form of the gene has been positively selected in non-Africans. The new study quantifies the advantage that the inactive form ("pseudogene") conferrred at 0.9% and times it to just before the most recent Out-of-Africa migration.
PLoS Biology Volume 4 | Issue 3 | MARCH 2006
Gene Losses during Human Origins
Xiaoxia Wang et al.
Pseudogenization is a widespread phenomenon in genome evolution, and it has been proposed to serve as an engine of evolutionary change, especially during human origins (the “less-is-more” hypothesis). However, there has been no comprehensive analysis of human-specific pseudogenes. Furthermore, it is unclear whether pseudogenization itself can be selectively favored and thus play an active role in human evolution. Here we conduct a comparative genomic analysis and a literature survey to identify 80 nonprocessed pseudogenes that were inactivated in the human lineage after its separation from the chimpanzee lineage. Many functions are involved among these genes, with chemoreception and immune response being outstandingly overrepresented, suggesting potential species-specific features in these aspects of human physiology. To explore the possibility of adaptive pseudogenization, we focus on CASPASE12, a cysteinyl aspartate proteinase participating in inflammatory and innate immune response to endotoxins. We provide population genetic evidence that the nearly complete fixation of a null allele at CASPASE12 has been driven by positive selection, probably because the null allele confers protection from severe sepsis. We estimate that the selective advantage of the null allele is about 0.9% and the pseudogenization started shortly before the out-of-Africa migration of modern humans. Interestingly, two other genes related to sepsis were also pseudogenized in humans, possibly by selection. These adaptive gene losses might have occurred because of changes in our environment or genetic background that altered the threat from or response to sepsis. The identification and analysis of human-specific pseudogenes open the door for understanding the roles of gene losses in human origins, and the demonstration that gene loss itself can be adaptive supports and extends the “less-is-more” hypothesis.
Link
PLoS Biology Volume 4 | Issue 3 | MARCH 2006
Gene Losses during Human Origins
Xiaoxia Wang et al.
Pseudogenization is a widespread phenomenon in genome evolution, and it has been proposed to serve as an engine of evolutionary change, especially during human origins (the “less-is-more” hypothesis). However, there has been no comprehensive analysis of human-specific pseudogenes. Furthermore, it is unclear whether pseudogenization itself can be selectively favored and thus play an active role in human evolution. Here we conduct a comparative genomic analysis and a literature survey to identify 80 nonprocessed pseudogenes that were inactivated in the human lineage after its separation from the chimpanzee lineage. Many functions are involved among these genes, with chemoreception and immune response being outstandingly overrepresented, suggesting potential species-specific features in these aspects of human physiology. To explore the possibility of adaptive pseudogenization, we focus on CASPASE12, a cysteinyl aspartate proteinase participating in inflammatory and innate immune response to endotoxins. We provide population genetic evidence that the nearly complete fixation of a null allele at CASPASE12 has been driven by positive selection, probably because the null allele confers protection from severe sepsis. We estimate that the selective advantage of the null allele is about 0.9% and the pseudogenization started shortly before the out-of-Africa migration of modern humans. Interestingly, two other genes related to sepsis were also pseudogenized in humans, possibly by selection. These adaptive gene losses might have occurred because of changes in our environment or genetic background that altered the threat from or response to sepsis. The identification and analysis of human-specific pseudogenes open the door for understanding the roles of gene losses in human origins, and the demonstration that gene loss itself can be adaptive supports and extends the “less-is-more” hypothesis.
Link
Y-chromosomes of Eastern India
This paper seems to take an opposite position compared to the other one by the same authors. From the paper:
Am J Phys Anthropol. 2006 Feb 16; [Epub ahead of print]
Phylogeography of mitochondrial DNA and Y-Chromosome haplogroups reveal asymmetric gene flow in populations of Eastern India.
Sahoo S, Kashyap VK.
Polymorphisms in mitochondrial (mt) DNA and Y-chromosomes of seven socially and linguistically diverse castes and tribes of Eastern India were examined to determine their genetic relationships, their origin, and the influence of demographic factors on population structure. Samples from the Orissa Brahmin, Karan, Khandayat, Gope, Juang, Saora, and Paroja were analyzed for mtDNA hypervariable sequence (HVS) I and II, eight Y-chromosome short tandem repeats (Y-STRs), and lineage-defining mutations diagnostic for Indian- and Eurasian-specific haplogroups. Our results reveal that haplotype diversity and mean pairwise differences (MPD) was higher in caste groups of the region (>0.998, for both systems) compared to tribes (0.917-0.996 for Y-STRs, and 0.958-0.988 for mtDNA haplotypes). The majority of paternal lineages belong to the R1a1, O2a, and H haplogroups (62.7%), while 73.2% of maternal lineages comprise the Indian-specific M*, M5, M30, and R* mtDNA haplogroups, with a sporadic occurrence of West Eurasian lineages. Our study reveals that Orissa Brahmins (a higher caste population) have a genetic affinity with Indo-European speakers of Eastern Europe, although the Y-chromosome data show that the genetic distances of populations are not correlated to their position in the caste hierarchy. The high frequency of the O2a haplogroup and absence of East Asian-specific mtDNA lineages in the Juang and Saora suggest that a migration of Austro-Asiatic tribes to mainland India was exclusively male-mediated which occurred during the demographic expansion of Neolithic farmers in southern China. The phylogeographic analysis of mtDNA and Y-chromosomes revealed varied ancestral sources for the diverse genetic components of the populations of Eastern India.
Link
Haplogroups H* and R2 were the two most common paternal lineages found in Orissa, although the frequency varied among populations. Caste populations displayed clear imprints of M17 (R1a1) at 38.5% and M172 (J2) at 5.7%, which are also found in high frequency in Eastern Europe and Central Asia (Semino et al., 2000; Underhill et al., 2000; Wells et al., 2001) and in the Middle East (Quintana-Murci et al., 2001), respectively. Among the Austro-Asiatic tribal groups, all the Juang Y-chromosomes exhibited the O2a lineage, which is common in eastern Asia (Karafet et al., 2001), while the Saora harbored lineages consisting of the F*, H, O2a, and P* haplogroups. The C haplogroup, which was associated with an early coastal migration of modern humans (Underhill et al., 2001), was found at a low frequency (1.6%) in the Paroja, and was completely absent in Austro-Asiatic tribes and the caste populations.As I have argued in my review of Indian Y-chromosome variation before, the origin of M17-related Y-chromosomes is not an either-or proposition between Central Asia and India, but rather it is possible that a group of such chromosomes are of Paleolithic antiquity in India, while a subset may have been of more recent introduction. No doubt, more light will be shed on this matter once the phylogeny of M17 is better developed.
Am J Phys Anthropol. 2006 Feb 16; [Epub ahead of print]
Phylogeography of mitochondrial DNA and Y-Chromosome haplogroups reveal asymmetric gene flow in populations of Eastern India.
Sahoo S, Kashyap VK.
Polymorphisms in mitochondrial (mt) DNA and Y-chromosomes of seven socially and linguistically diverse castes and tribes of Eastern India were examined to determine their genetic relationships, their origin, and the influence of demographic factors on population structure. Samples from the Orissa Brahmin, Karan, Khandayat, Gope, Juang, Saora, and Paroja were analyzed for mtDNA hypervariable sequence (HVS) I and II, eight Y-chromosome short tandem repeats (Y-STRs), and lineage-defining mutations diagnostic for Indian- and Eurasian-specific haplogroups. Our results reveal that haplotype diversity and mean pairwise differences (MPD) was higher in caste groups of the region (>0.998, for both systems) compared to tribes (0.917-0.996 for Y-STRs, and 0.958-0.988 for mtDNA haplotypes). The majority of paternal lineages belong to the R1a1, O2a, and H haplogroups (62.7%), while 73.2% of maternal lineages comprise the Indian-specific M*, M5, M30, and R* mtDNA haplogroups, with a sporadic occurrence of West Eurasian lineages. Our study reveals that Orissa Brahmins (a higher caste population) have a genetic affinity with Indo-European speakers of Eastern Europe, although the Y-chromosome data show that the genetic distances of populations are not correlated to their position in the caste hierarchy. The high frequency of the O2a haplogroup and absence of East Asian-specific mtDNA lineages in the Juang and Saora suggest that a migration of Austro-Asiatic tribes to mainland India was exclusively male-mediated which occurred during the demographic expansion of Neolithic farmers in southern China. The phylogeographic analysis of mtDNA and Y-chromosomes revealed varied ancestral sources for the diverse genetic components of the populations of Eastern India.
Link
February 18, 2006
mtDNA of Ancient Peruvians
One more for the Ancient DNA Compendium
Am J Phys Anthropol. 2006 Feb 16; [Epub ahead of print]
Mitochondrial DNA analysis of ancient Peruvian highlanders.
Shinoda KI, Adachi N, Guillen S, Shimada I.
Ancient DNA recovered from 57 individuals excavated by Hiram Bingham at the rural communities of Paucarcancha, Patallacta, and Huata near the famed Inca royal estate and ritual site of Machu Picchu was analyzed by polymerase chain reaction, and the results were compared with ancient and modern DNA from various Central Andean areas to test their hypothesized indigenous highland origins. The control and coding regions of the mitochondrial DNA (mtDNA) of 35 individuals in this group were sequenced, and the haplogroups of each individual were determined. The frequency data for the haplogroups of these samples show clear proximity to those of modern Quechua and Aymara populations in the Peruvian and Bolivian highlands, and contrast with those of pre-Hispanic individuals of the north coast of Peru that we defined previously. Our study suggests a strong genetic affinity between sampled late pre-Hispanic individuals and modern Andean highlanders. A previous analysis of the Machu Picchu osteological collection suggests that the residents there were a mixed group of natives from various coastal and highland regions relocated by the Inca state for varied purposes. Overall, our study indicates that the sampled individuals from Paucarcancha and Patallacta were indigenous highlanders who provided supportive roles for nearby Machu Picchu.
Link
Am J Phys Anthropol. 2006 Feb 16; [Epub ahead of print]
Mitochondrial DNA analysis of ancient Peruvian highlanders.
Shinoda KI, Adachi N, Guillen S, Shimada I.
Ancient DNA recovered from 57 individuals excavated by Hiram Bingham at the rural communities of Paucarcancha, Patallacta, and Huata near the famed Inca royal estate and ritual site of Machu Picchu was analyzed by polymerase chain reaction, and the results were compared with ancient and modern DNA from various Central Andean areas to test their hypothesized indigenous highland origins. The control and coding regions of the mitochondrial DNA (mtDNA) of 35 individuals in this group were sequenced, and the haplogroups of each individual were determined. The frequency data for the haplogroups of these samples show clear proximity to those of modern Quechua and Aymara populations in the Peruvian and Bolivian highlands, and contrast with those of pre-Hispanic individuals of the north coast of Peru that we defined previously. Our study suggests a strong genetic affinity between sampled late pre-Hispanic individuals and modern Andean highlanders. A previous analysis of the Machu Picchu osteological collection suggests that the residents there were a mixed group of natives from various coastal and highland regions relocated by the Inca state for varied purposes. Overall, our study indicates that the sampled individuals from Paucarcancha and Patallacta were indigenous highlanders who provided supportive roles for nearby Machu Picchu.
Link
February 17, 2006
Documenting domestication: the intersection of genetics and archaeology
A pretty good review of the archaeology and genetics of domesticated plants and animals.
Trends in Genetics (Article in Press)
Documenting domestication: the intersection of genetics and archaeology
Melinda A. Zeder et al.
Domestication, a process of increasing mutual dependence between human societies and the plant and animal populations they target, has long been an area of interest in genetics and archaeology. Geneticists seek out markers of domestication in the genomes of domesticated species, both past and present day. Archaeologists examine the archaeological record for complementary markers – evidence of the human behavior patterns that cause the genetic changes associated with domestication, and the morphological changes in target species that result from them. In this article, we summarize the recent advances in genetics and archaeology in documenting plant and animal domestication, and highlight several promising areas where the complementary perspectives of both disciplines provide reciprocal illumination.
Link
Trends in Genetics (Article in Press)
Documenting domestication: the intersection of genetics and archaeology
Melinda A. Zeder et al.
Domestication, a process of increasing mutual dependence between human societies and the plant and animal populations they target, has long been an area of interest in genetics and archaeology. Geneticists seek out markers of domestication in the genomes of domesticated species, both past and present day. Archaeologists examine the archaeological record for complementary markers – evidence of the human behavior patterns that cause the genetic changes associated with domestication, and the morphological changes in target species that result from them. In this article, we summarize the recent advances in genetics and archaeology in documenting plant and animal domestication, and highlight several promising areas where the complementary perspectives of both disciplines provide reciprocal illumination.
Link
Quantitative trait loci for intelligence
It appears that the hunt for genes affecting intelligence is not going well. I can't say that I'm surprised, because I have always maintained that intelligence is an emergent property of a set of co-operating genes during development in a particular environment and I don't anticipate that the geno-centric approach will take us closer to understanding it.
Intelligence, and -I believe- other complex traits are like complex dishes with many ingredients. The ingredients themselves (e.g., salt, lettuce, or chicken) are themselves unremarkable, but it is the way that they are put together and turned on and off by internal and external stimuli (the pot, the temperature, time, etc.) that makes a good dish.
Intelligence (Article in Press)
The quest for quantitative trait loci associated with intelligence
Robert Plomin et al.
Abstract
Progress towards identifying quantitative trait loci (QTLs) for complex traits like intelligence and common disorders like mental retardation has been slower than expected. An important factor is that most QTL effects may be much smaller than expected—not just 1% effect sizes but perhaps effects as small as .1%. If so, this would mean that studies have been seriously underpowered to detect and to replicate QTL effects. We have used microarrays to genotype DNA pooled for groups of low versus high intelligence in order to screen very large numbers of single nucleotide polymorphisms (SNPs) on very large samples in the quest for QTLs of very small effect size: We find no effect sizes greater than .5%. Microarrays with 500,000 SNPs are now available that facilitate genomewide scans which will make it possible to identify nearly all SNP associations that account for 1% of the variance of intelligence—if there are any QTL effect sizes as large as 1%.
Link
Intelligence, and -I believe- other complex traits are like complex dishes with many ingredients. The ingredients themselves (e.g., salt, lettuce, or chicken) are themselves unremarkable, but it is the way that they are put together and turned on and off by internal and external stimuli (the pot, the temperature, time, etc.) that makes a good dish.
Intelligence (Article in Press)
The quest for quantitative trait loci associated with intelligence
Robert Plomin et al.
Abstract
Progress towards identifying quantitative trait loci (QTLs) for complex traits like intelligence and common disorders like mental retardation has been slower than expected. An important factor is that most QTL effects may be much smaller than expected—not just 1% effect sizes but perhaps effects as small as .1%. If so, this would mean that studies have been seriously underpowered to detect and to replicate QTL effects. We have used microarrays to genotype DNA pooled for groups of low versus high intelligence in order to screen very large numbers of single nucleotide polymorphisms (SNPs) on very large samples in the quest for QTLs of very small effect size: We find no effect sizes greater than .5%. Microarrays with 500,000 SNPs are now available that facilitate genomewide scans which will make it possible to identify nearly all SNP associations that account for 1% of the variance of intelligence—if there are any QTL effect sizes as large as 1%.
Link
Sex-biased migration in humans
Bioessays. 2006 Feb 14;28(3):290-300 [Epub ahead of print]
Sex-biased migration in humans: what should we expect from genetic data?
Wilkins JF, Marlowe FW.
Different patterns of mitochondrial and Y-chromosome diversity have been cited as evidence of long-term patrilocality in human populations. However, what patterns are expected depends on the nature of the sampling scheme. Samples from a local region reveal only the recent demographic history of that region, whereas sampling over larger geographic scales accesses older demographic processes. A historical change in migration becomes evident first at local geographic scales, and alters global patterns of genetic diversity only after sufficient time has passed. Analysis of forager populations in the ethnographic record suggests that patrilocality may not have predominated among pre-agricultural humans. The higher female migration rate inferred by some genetic studies may reflect a shift to patrilocality in association with the emergence of agriculture. A recent global survey does not show the expected effects of higher female migration, possibly because the sampling scheme used for this study is accessing pre-agricultural human migration patterns. In this paper, we show how the demographic shift associated with agriculture might affect genetic diversity over different spatial scales. We also consider the prospects for studying sex-biased migration using the X-linked and autosomal markers. These multi-locus comparisons have the potential of providing more robust estimates of sex differences than Y-linked and mitochondrial data, but only if a very large number of loci are included in the analysis. BioEssays 28: 290-300, 2006. (c) 2006 Wiley Periodicals, Inc.
Link
Sex-biased migration in humans: what should we expect from genetic data?
Wilkins JF, Marlowe FW.
Different patterns of mitochondrial and Y-chromosome diversity have been cited as evidence of long-term patrilocality in human populations. However, what patterns are expected depends on the nature of the sampling scheme. Samples from a local region reveal only the recent demographic history of that region, whereas sampling over larger geographic scales accesses older demographic processes. A historical change in migration becomes evident first at local geographic scales, and alters global patterns of genetic diversity only after sufficient time has passed. Analysis of forager populations in the ethnographic record suggests that patrilocality may not have predominated among pre-agricultural humans. The higher female migration rate inferred by some genetic studies may reflect a shift to patrilocality in association with the emergence of agriculture. A recent global survey does not show the expected effects of higher female migration, possibly because the sampling scheme used for this study is accessing pre-agricultural human migration patterns. In this paper, we show how the demographic shift associated with agriculture might affect genetic diversity over different spatial scales. We also consider the prospects for studying sex-biased migration using the X-linked and autosomal markers. These multi-locus comparisons have the potential of providing more robust estimates of sex differences than Y-linked and mitochondrial data, but only if a very large number of loci are included in the analysis. BioEssays 28: 290-300, 2006. (c) 2006 Wiley Periodicals, Inc.
Link
February 16, 2006
Co-operation in structured heterogeneous populations
Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0508201103
Evolutionary dynamics of social dilemmas in structured heterogeneous populations
F. C. Santos et al.
Real populations have been shown to be heterogeneous, in which some individuals have many more contacts than others. This fact contrasts with the traditional homogeneous setting used in studies of evolutionary game dynamics. We incorporate heterogeneity in the population by studying games on graphs, in which the variability in connectivity ranges from single-scale graphs, for which heterogeneity is small and associated degree distributions exhibit a Gaussian tale, to scale-free graphs, for which heterogeneity is large with degree distributions exhibiting a power-law behavior. We study the evolution of cooperation, modeled in terms of the most popular dilemmas of cooperation. We show that, for all dilemmas, increasing heterogeneity favors the emergence of cooperation, such that long-term cooperative behavior easily resists short-term noncooperative behavior. Moreover, we show how cooperation depends on the intricate ties between individuals in scale-free populations.
Link
Evolutionary dynamics of social dilemmas in structured heterogeneous populations
F. C. Santos et al.
Real populations have been shown to be heterogeneous, in which some individuals have many more contacts than others. This fact contrasts with the traditional homogeneous setting used in studies of evolutionary game dynamics. We incorporate heterogeneity in the population by studying games on graphs, in which the variability in connectivity ranges from single-scale graphs, for which heterogeneity is small and associated degree distributions exhibit a Gaussian tale, to scale-free graphs, for which heterogeneity is large with degree distributions exhibiting a power-law behavior. We study the evolution of cooperation, modeled in terms of the most popular dilemmas of cooperation. We show that, for all dilemmas, increasing heterogeneity favors the emergence of cooperation, such that long-term cooperative behavior easily resists short-term noncooperative behavior. Moreover, we show how cooperation depends on the intricate ties between individuals in scale-free populations.
Link
February 15, 2006
Teens made Paleolithic cave art (?)
FAIRBANKS, Alaska -- Long accustomed to lifting mammoth bones from mudbanks and museum shelves and making sketches from cave art to gather details about Pleistocene animal anatomy, renowned paleobiologist and artist R. Dale Guthrie offers a fascinating and controversial interpretation of ancient cave art in his new book “The Nature of Paleolithic Art.”
This ancient art was made during the late Pleistocene, about 10,000 to 35,000 years ago, and has typically been the purview of art historians and anthropologists, many of whom view Paleolithic art as done by accomplished shaman-artists. “This assumption may be true of a few of the best known and better-drawn images, but these are a small proportion of preserved Paleolithic art,” Guthrie said.
Using new forensic techniques on fossil handprints of the artists and examining thousands of images, “I found that all ages and both sexes were making art, not just the senior male shamans,” Guthrie said. These included hundreds of prints made as ocher, manganese, or clay negatives and a few positive prints made with pigments or mud applied to hands that were then placed on cave surfaces.
“The possibility that adolescent giggles and snickers may have echoed in dark cave passages as often as the rhythm of a shaman’s chant demeans neither artists nor art,” writes Guthrie.
“I was using Paleolithic art both to appreciate the colorful renditions and to find useful and interesting details about Pleistocene animal anatomy,” said Guthrie, professor emeritus of the Institute of Arctic Biology at the University of Alaska Fairbanks. A symposium of Paleolithic art scholars in 1979 “... set me on a new course of trying to place Paleolithic art in a larger dimension of natural history and linking artistic behavior to our evolutionary past,” writes Guthrie.
The book, which contains more than 3,000 images all drawn by Guthrie, is about more than art. It’s about good parenting, children, romantic love, lust, play, graffiti, risk-proneness, missing shields, hour-glass figures, striped horses, seas of grasses, and cold dry winds – it’s about life on the margins of the Ice Age Mammoth Steppe.
LinkAn utter refutation of the 'Fundamental Theorem of the HapMap'
This sounds potentially very important, but it will take some reading before I can make sense of it. Interestingly, the first author has five different institutional affiliations, which has to be some kind of record...
Read also HapMap publication explosion.
European Journal of Human Genetics (online early)
An utter refutation of the 'Fundamental Theorem of the HapMap'
Joseph D Terwilliger and Tero Hiekkalinna
Abstract
The International HapMap Project was proposed in order to quantify linkage disequilibrium (LD) relationships among human DNA polymorphisms in an assortment of populations, in order to facilitate the process of selecting a minimal set of markers that could capture most of the signal from the untyped markers in a genome-wide association study. The central dogma can be summarized by the argument that if a marker is in tight LD with a polymorphism that directly impacts disease risk, as measured by the metric r2, then one would be able to detect an association between the marker and disease with sample size that was increased by a factor of 1/r2 over that needed to detect the effect of the functional variant directly. This 'fundamental theorem' holds, however, only if one assumes that the LD between loci and the etiological effect of the functional variant are independent of each other, that they are statistically independent of all other etiological factors (in exposure and action), that sampling is prospective, and that the estimates of r2 are accurate. None of these are standard operating assumptions, however. We describe the ramifications of these implicit assumptions, and provide simple examples in which the effects of a functional variant could be unequivocally detected if it were directly genotyped, even as markers in high LD with the functional variant would never show association with disease, even in infinite sample sizes. Both theoretical and empirical refutation of the central dogma of genome-wide association studies is thus presented.
Link
Read also HapMap publication explosion.
European Journal of Human Genetics (online early)
An utter refutation of the 'Fundamental Theorem of the HapMap'
Joseph D Terwilliger and Tero Hiekkalinna
Abstract
The International HapMap Project was proposed in order to quantify linkage disequilibrium (LD) relationships among human DNA polymorphisms in an assortment of populations, in order to facilitate the process of selecting a minimal set of markers that could capture most of the signal from the untyped markers in a genome-wide association study. The central dogma can be summarized by the argument that if a marker is in tight LD with a polymorphism that directly impacts disease risk, as measured by the metric r2, then one would be able to detect an association between the marker and disease with sample size that was increased by a factor of 1/r2 over that needed to detect the effect of the functional variant directly. This 'fundamental theorem' holds, however, only if one assumes that the LD between loci and the etiological effect of the functional variant are independent of each other, that they are statistically independent of all other etiological factors (in exposure and action), that sampling is prospective, and that the estimates of r2 are accurate. None of these are standard operating assumptions, however. We describe the ramifications of these implicit assumptions, and provide simple examples in which the effects of a functional variant could be unequivocally detected if it were directly genotyped, even as markers in high LD with the functional variant would never show association with disease, even in infinite sample sizes. Both theoretical and empirical refutation of the central dogma of genome-wide association studies is thus presented.
Link
More on genetic ancestry with 10,000+ SNPs (or just 10)
See previous post here.
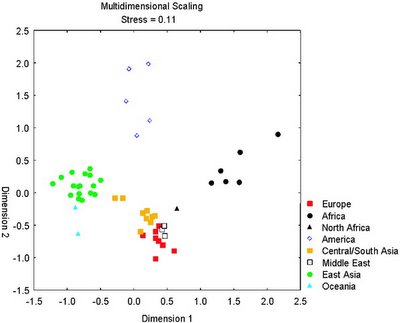
A very exciting new paper has just appeared in AJHG. The authors have used the 10,000+ SNP array by Affymetrix to study the individuals from the Y chromosome consortium panel. Note that these individuals were originally collected to study Y chromosomal variation, but in this case, they were studied because they represent a globally diverse set of people, and their genomic variation was studied.
The researchers were able to discern the population origin of these 76 individuals using just 10 SNPs. So, it only takes 10 nucleotides to infer whether someone is Sub-Saharan African, West Eurasian, East Asian, or Native American.
Of course, this ability could be due to overfitting in the small 76-individual sample. So, they tested against the larger 1000+ individual CEPH panel which includes 50+ populations from around the world, and were able to correctly infer the ancestry of all individuals.
Thus, it is demonstrated that a very small set of carefully selected polymorphisms are enough to discover the continental origin of an individual:
Finally, the authors also asked why the very informative SNPs show such high frequency differences in the studied populations:
Am. J. Hum. Genet. (online early)
Proportioning Whole-Genome Single-Nucleotide–Polymorphism Diversity for the Identification of Geographic Population Structure and Genetic Ancestry
Oscar Lao et al.
The identification of geographic population structure and genetic ancestry on the basis of a minimal set of genetic markers is desirable for a wide range of applications in medical and forensic sciences. However, the absence of sharp discontinuities in the neutral genetic diversity among human populations implies that, in practice, a large number of neutral markers will be required to identify the genetic ancestry of one individual. We showed that it is possible to reduce the amount of markers required for detecting continental population structure to only 10 single-nucleotide polymorphisms (SNPs), by applying a newly developed ascertainment algorithm to Affymetrix GeneChip Mapping 10K SNP array data that we obtained from samples of globally dispersed human individuals (the Y Chromosome Consortium panel). Furthermore, this set of SNPs was able to recover the genetic ancestry of individuals from all four continents represented in the original data set when applied to an independent, much larger, worldwide population data set (Centre d'Etude du Polymorphisme Humain–Human Genome Diversity Project Cell Line Panel). Finally, we provide evidence that the unusual patterns of genetic variation we observed at the respective genomic regions surrounding the five most informative SNPs is in agreement with local positive selection being the explanation for the striking SNP allele-frequency differences we found between continental groups of human populations
Link
A very exciting new paper has just appeared in AJHG. The authors have used the 10,000+ SNP array by Affymetrix to study the individuals from the Y chromosome consortium panel. Note that these individuals were originally collected to study Y chromosomal variation, but in this case, they were studied because they represent a globally diverse set of people, and their genomic variation was studied.
The researchers were able to discern the population origin of these 76 individuals using just 10 SNPs. So, it only takes 10 nucleotides to infer whether someone is Sub-Saharan African, West Eurasian, East Asian, or Native American.
Of course, this ability could be due to overfitting in the small 76-individual sample. So, they tested against the larger 1000+ individual CEPH panel which includes 50+ populations from around the world, and were able to correctly infer the ancestry of all individuals.
Thus, it is demonstrated that a very small set of carefully selected polymorphisms are enough to discover the continental origin of an individual:

Finally, the authors also asked why the very informative SNPs show such high frequency differences in the studied populations:
Thus, although the SNPs from the whole-genome analyses used to identify ancestry-informative markers were noncoding, our data indicate that the significant population differences of the markers with maximum informativeness of ancestry seem to be shaped by positive selection rather than by genetic drift.
Am. J. Hum. Genet. (online early)
Proportioning Whole-Genome Single-Nucleotide–Polymorphism Diversity for the Identification of Geographic Population Structure and Genetic Ancestry
Oscar Lao et al.
The identification of geographic population structure and genetic ancestry on the basis of a minimal set of genetic markers is desirable for a wide range of applications in medical and forensic sciences. However, the absence of sharp discontinuities in the neutral genetic diversity among human populations implies that, in practice, a large number of neutral markers will be required to identify the genetic ancestry of one individual. We showed that it is possible to reduce the amount of markers required for detecting continental population structure to only 10 single-nucleotide polymorphisms (SNPs), by applying a newly developed ascertainment algorithm to Affymetrix GeneChip Mapping 10K SNP array data that we obtained from samples of globally dispersed human individuals (the Y Chromosome Consortium panel). Furthermore, this set of SNPs was able to recover the genetic ancestry of individuals from all four continents represented in the original data set when applied to an independent, much larger, worldwide population data set (Centre d'Etude du Polymorphisme Humain–Human Genome Diversity Project Cell Line Panel). Finally, we provide evidence that the unusual patterns of genetic variation we observed at the respective genomic regions surrounding the five most informative SNPs is in agreement with local positive selection being the explanation for the striking SNP allele-frequency differences we found between continental groups of human populations
Link
February 14, 2006
Anatomically modern humans
I think that this definition by Lahr & Wright is useful enough as a reference, augmented with links explaining various terms:
Lahr, M.M. & Wright, R.S.V. (1996) The question of robusticity and the relationship between cranial size and shape in Homo sapiens. Journal of Human Evolution, 31: 157-191.
...it is generally agreed that a modern skull should present a relatively small face tucked under the vault that is relatively short and high, a relatively vertical forehead, parietal enlargement, a relatively rounded occiput, a flexed cranial base, a canine fossa, an occipital protuberance in the occipital bone and mental eminence or chin. Most descriptions of modern H. sapiens would also include skeletal gracility as characterizing the group.But read the Modernity mess as well by Wolpoff & Caspari.
Lahr, M.M. & Wright, R.S.V. (1996) The question of robusticity and the relationship between cranial size and shape in Homo sapiens. Journal of Human Evolution, 31: 157-191.
February 11, 2006
The second search for Peking Man
Many cranial fossils have strange histories. Some, like Piltdown Man and Galley Hill and Vogelherd persist for decades as important prehistoric remains, even though later dating reveals them to be much more recent than originally thought. Others like Kennewick Man become objects of a legal battle as to whether or not they should be buried and lost for science, or studied. And there are others like Combe Capelle who was lost during World War II, and then found decades later.
There are casts of all important skulls, so the loss of a specimen does not mean that it can no longer be studied by anthropologists. Unfortunately, newer techniques involving the extraction of DNA from bones only work with real skulls; hence, the urgency to rediscover misplaced fossils.
The Peking Man was lost during World War II as well. Thankfully, Franz Weidenreich had made casts of the skulls only years after they had been brought to the ground, and these casts have played a key role in subsequent discussions on human origins.
Now, a news story hints that Peking Man may not have been lost forever:
Let's hope they succeed.
There are casts of all important skulls, so the loss of a specimen does not mean that it can no longer be studied by anthropologists. Unfortunately, newer techniques involving the extraction of DNA from bones only work with real skulls; hence, the urgency to rediscover misplaced fossils.
The Peking Man was lost during World War II as well. Thankfully, Franz Weidenreich had made casts of the skulls only years after they had been brought to the ground, and these casts have played a key role in subsequent discussions on human origins.
Now, a news story hints that Peking Man may not have been lost forever:
ZHOUKOUDIAN, CHINA -- For more than two decades, Yang Shoukai had hoarded his secret, unsure what to do with a possible clue to one of China's most baffling mysteries.
As construction supervisor on the site of an abandoned U.S. military barracks in Tianjin in 1982, he had discovered a strange cement box in the basement of the old wartime barracks. He tried to dig it up, but lacked the proper tools, and the box was buried under a new medical laboratory.
Mr. Yang, now retired and in poor health, is convinced that the box contains a 500,000-year-old archeological treasure that China has hunted for in vain since the Second World War: the missing skulls of Peking Man, one of the most famous links in the evolution of prehistoric humans.
...
Chinese specialists have conducted many failed searches for the skulls, but the new committee is optimistic. "I think we will find them," said Dong Cuiping, vice-director of the Peking Man Museum.
"Since 1941, the searches were always done by non-governmental people. Now this work is on the government's agenda. We're drawing more attention from society, and more clues - not only from China but outside China too."
If the skulls are recovered, they could become the basis for a major breakthrough in the understanding of human evolution, she said.
"When we first found the skulls, our science was not very advanced. Now we have much better technology. If we recover them, we could make new scientific discoveries."
Let's hope they succeed.
Lucky find reveals biggest ancient cave
Link (Kathimerini)
A farmer in northern Greece has stumbled across a 2,300-year-old chiseled cave with eight chambers and measuring some 63 square meters — the biggest ever discovered in this country — it was revealed yesterday.
The cave was found near the ancient city of Pella, which was the capital of the Macedonian kingdom. Archaeologists are studying the cave and believe it was probably used as a tomb by a wealthy Macedonian family between the second and third centuries BC, the Athens News Agency reported.
Two gold earrings, several bronze coins, three marble funerary stelae and a number of ceramic vessels were found inside the cave.
Previously, the largest chiseled cave found in Greece had three chambers. The cave near Pella has partly retained its original wall coloring of red, sky blue and gold.
February 10, 2006
The shape of things to come in autosomal DNA analysis
In the recent African American Lives documentary, geneticists were able to estimate the most probable African origin of Henry Louis Gates, Jr. by using a 11,555-SNP array. The following paper reports on the study of several human populations using this array.
Until now, it was well known that major continental origin could be predicted accurately, but by increasing the number of polymorphisms used, one can go one step further and discover patterns at the intra-continental level.
The following plot of the first PCA components is pretty self-explanatory. As you can see, very clear clusters corresponding to populations emerge when such large numbers of polymorphisms are used.

Human Genomics Volume 2, Number 2, June 2005
Large-scale SNP analysis reveals clustered and continuous patterns of human genetic variation
Shriver, Mark D et al.
Abstract:
Understanding the distribution of human genetic variation is an important foundation for research into the genetics of common diseases. Some of the alleles that modify common disease risk are themselves likely to be common and, thus, amenable to identification using gene-association methods. A problem with this approach is that the large sample sizes required for sufficient statistical power to detect alleles with moderate effect make gene-association studies susceptible to false-positive findings as the result of population stratification.1,2 Such type I errors can be eliminated by using either family-based association tests or methods that sufficiently adjust for population stratification.3–5 These methods require the availability of genetic markers that can detect and, thus, control for sources of genetic stratification among populations. In an effort to investigate population stratification and identify appropriate marker panels, we have analysed 11,555 single nucleotide polymorphisms in 203 individuals from 12 diverse human populations. Individuals in each population cluster to the exclusion of individuals from other populations using two clustering methods. Higher-order branching and clustering of the populations are consistent with the geographic origins of populations and with previously published genetic analyses. These data provide a valuable resource for the definition of marker panels to detect and control for population stratification in population-based gene identification studies. Using three US resident populations (European-American, African-American and Puerto Rican), we demonstrate how such studies can proceed, quantifying proportional ancestry levels and detecting significant admixture structure in each of these populations.
Link
Until now, it was well known that major continental origin could be predicted accurately, but by increasing the number of polymorphisms used, one can go one step further and discover patterns at the intra-continental level.
The following plot of the first PCA components is pretty self-explanatory. As you can see, very clear clusters corresponding to populations emerge when such large numbers of polymorphisms are used.

Human Genomics Volume 2, Number 2, June 2005
Large-scale SNP analysis reveals clustered and continuous patterns of human genetic variation
Shriver, Mark D et al.
Abstract:
Understanding the distribution of human genetic variation is an important foundation for research into the genetics of common diseases. Some of the alleles that modify common disease risk are themselves likely to be common and, thus, amenable to identification using gene-association methods. A problem with this approach is that the large sample sizes required for sufficient statistical power to detect alleles with moderate effect make gene-association studies susceptible to false-positive findings as the result of population stratification.1,2 Such type I errors can be eliminated by using either family-based association tests or methods that sufficiently adjust for population stratification.3–5 These methods require the availability of genetic markers that can detect and, thus, control for sources of genetic stratification among populations. In an effort to investigate population stratification and identify appropriate marker panels, we have analysed 11,555 single nucleotide polymorphisms in 203 individuals from 12 diverse human populations. Individuals in each population cluster to the exclusion of individuals from other populations using two clustering methods. Higher-order branching and clustering of the populations are consistent with the geographic origins of populations and with previously published genetic analyses. These data provide a valuable resource for the definition of marker panels to detect and control for population stratification in population-based gene identification studies. Using three US resident populations (European-American, African-American and Puerto Rican), we demonstrate how such studies can proceed, quantifying proportional ancestry levels and detecting significant admixture structure in each of these populations.
Link
Behavioral Inhibition and Blond Hair
Via Gene Expression.
Biological Psychology (Article in Press)
Association of behavioral inhibition with hair pigmentation in a European sample
Eva Moehler et al.
Abstract
Behavioral inhibition, a temperamental trait signalling a predisposition to childhood and adolescent anxiety disorders, is slightly more frequent in America among Caucasian children having blue irises. This paper examines a community sample of 101 German toddlers assessed for behavioral inhibition in a standardized laboratory procedure. Hair pigmentation was found to be significantly associated with behavioral inhibition in the sense that blond children exhibited higher fear scores. As in American samples, blue-eyed children had a higher fear score than did other children, but this difference was not statistically significant.
Link
Biological Psychology (Article in Press)
Association of behavioral inhibition with hair pigmentation in a European sample
Eva Moehler et al.
Abstract
Behavioral inhibition, a temperamental trait signalling a predisposition to childhood and adolescent anxiety disorders, is slightly more frequent in America among Caucasian children having blue irises. This paper examines a community sample of 101 German toddlers assessed for behavioral inhibition in a standardized laboratory procedure. Hair pigmentation was found to be significantly associated with behavioral inhibition in the sense that blond children exhibited higher fear scores. As in American samples, blue-eyed children had a higher fear score than did other children, but this difference was not statistically significant.
Link
February 09, 2006
Ethnicity-specific disease risk in African Americans
This paper shows how a particular gene variant was introduced to the African American population by admixture with Europeans and how African Americans are at a higher risk of heart disease because of it.
This illustrates how a gene that has originated in one population can lead to problems when it is found in a different genetic-environmental context.
Nature Genetics 38, 68 - 74 (2006)
Published online: 10 November 2005; | doi:10.1038/ng1692
A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction
Anna Helgadottir et al.
Variants of the gene ALOX5AP (also known as FLAP) encoding arachidonate 5-lipoxygenase activating protein are known to be associated with risk of myocardial infarction1. Here we show that a haplotype (HapK) spanning the LTA4H gene encoding leukotriene A4 hydrolase, a protein in the same biochemical pathway as ALOX5AP, confers modest risk of myocardial infarction in an Icelandic cohort. Measurements of leukotriene B4 (LTB4) production suggest that this risk is mediated through upregulation of the leukotriene pathway. Three cohorts from the United States also show that HapK confers a modest relative risk (1.16) in European Americans, but it confers a threefold larger risk in African Americans. About 27% of the European American controls carried at least one copy of HapK, as compared with only 6% of African American controls. Our analyses indicate that HapK is very rare in Africa and that its occurrence in African Americans is due to European admixture. Interactions with other genetic or environmental risk factors that are more common in African Americans are likely to account for the greater relative risk conferred by HapK in this group.
Link
More African American Lives
I watched African American Lives, finding it to be quite well done, and informative. I took some notes on some of the celebrities which were shown in the program.
Henry Louis Jr, 50%-50% European/Sub-Saharan African

Oprah Winfrey 89%-8%-3% Sub-Saharan African/Native American/East Asian

Quincy Jones 63%/34% Sub-Saharan African/European

Sarah Lawrence Lightfoot 45% European

Chris Tucker 83%/10% Sub-Saharan African/Native American (?)

Mae Jemison 13% East Asian

Whoopi Goldberg 90% Sub-Saharan African

Additionally, Mark Shriver was shown to take a 3D scan of Henry Louis, Jr's head, and it was said that he was working on correlating facial features with genetic admixture. On the minus side, the 13% "East Asian" component in Mae Jamison was explained by "Chinese laborers", rather than the more probable explanation of a difficulty in distinguishing between Native Americans and East Asians.
See also Mozart's Skull and the DNA of the famous
Henry Louis Jr, 50%-50% European/Sub-Saharan African

Oprah Winfrey 89%-8%-3% Sub-Saharan African/Native American/East Asian

Quincy Jones 63%/34% Sub-Saharan African/European

Sarah Lawrence Lightfoot 45% European

Chris Tucker 83%/10% Sub-Saharan African/Native American (?)

Mae Jemison 13% East Asian

Whoopi Goldberg 90% Sub-Saharan African

Additionally, Mark Shriver was shown to take a 3D scan of Henry Louis, Jr's head, and it was said that he was working on correlating facial features with genetic admixture. On the minus side, the 13% "East Asian" component in Mae Jamison was explained by "Chinese laborers", rather than the more probable explanation of a difficulty in distinguishing between Native Americans and East Asians.
See also Mozart's Skull and the DNA of the famous
February 07, 2006
The sex of attractive faces is identified more easily
Perception. 2005;34(12):1459-74.
The role of facial attractiveness and facial masculinity/femininity in sex classification of faces.
Hoss RA, Ramsey JL, Griffin AM, Langlois JH.
We tested whether adults (experiment 1) and 4 - 5-year-old children (experiment 2) identify the sex of highly attractive faces faster and more accurately than not very attractive faces in a reaction-time task. We also assessed whether facial masculinity/femininity facilitated identification of sex. Results showed that attractiveness facilitated adults' sex classification of both female and male faces and children's sex classification of female, but not male, faces. Moreover, attractiveness affected the speed and accuracy of sex classification independently of masculinity/femininity. High masculinity in male faces, but not high femininity in female faces, also facilitated sex classification for both adults and children. These findings provide important new data on how the facial cues of attractiveness and masculinity/femininity contribute to the task of sex classification and provide evidence for developmental differences in how adults and children use these cues. Additionally, these findings provide support for Langlois and Roggman's (1990 Psychological Science 1 115 121) averageness theory of attractiveness.
Link
Genetic influences on life span kick in after age 60
Human Genetics (Online first)
Genetic influence on human lifespan and longevity
Jacob vB. Hjelmborg et al.
Abstract There is an intense search for longevity genes in both animal models and humans. Human family studies have indicated that a modest amount of the overall variation in adult lifespan (approximately 20–30%) is accounted for by genetic factors. But it is not known if genetic factors become increasingly important for survival at the oldest ages. We study the genetic influence on human lifespan and how it varies with age using the almost extinct cohorts of Danish, Finnish and Swedish twins born between 1870 and 1910 comprising 20,502 individuals followed until 2003–2004. We first estimate mean lifespan of twins by lifespan of co-twin and then turn to the relative recurrence risk of surviving to a given age. Mean lifespan for male monozygotic (MZ) twins increases 0.39 [95% CI (0.28, 0.50)] years for every year his co-twin survives past age 60 years. This rate is significantly greater than the rate of 0.21 (0.11, 0.30) for dizygotic (DZ) males. Females and males have similar rates and these are negligible before age 60 for both MZ and DZ pairs. We moreover find that having a co-twin surviving to old ages substantially and significantly increases the chance of reaching the same old age and this chance is higher for MZ than for DZ twins. The relative recurrence risk of reaching age 92 is 4.8 (2.2, 7.5) for MZ males, which is significantly greater than the 1.8 (0.10, 3.4) for DZ males. The patterns for females and males are very similar, but with a shift of the female pattern with age that corresponds to the better female survival. Similar results arise when considering only those Nordic twins that survived past 75 years of age. The present large population based study shows genetic influence on human lifespan. While the estimated overall strength of genetic influence is compatible with previous studies, we find that genetic influences on lifespan are minimal prior to age 60 but increase thereafter. These findings provide a support for the search for genes affecting longevity in humans, especially at advanced ages.
Link
Genetic influence on human lifespan and longevity
Jacob vB. Hjelmborg et al.
Abstract There is an intense search for longevity genes in both animal models and humans. Human family studies have indicated that a modest amount of the overall variation in adult lifespan (approximately 20–30%) is accounted for by genetic factors. But it is not known if genetic factors become increasingly important for survival at the oldest ages. We study the genetic influence on human lifespan and how it varies with age using the almost extinct cohorts of Danish, Finnish and Swedish twins born between 1870 and 1910 comprising 20,502 individuals followed until 2003–2004. We first estimate mean lifespan of twins by lifespan of co-twin and then turn to the relative recurrence risk of surviving to a given age. Mean lifespan for male monozygotic (MZ) twins increases 0.39 [95% CI (0.28, 0.50)] years for every year his co-twin survives past age 60 years. This rate is significantly greater than the rate of 0.21 (0.11, 0.30) for dizygotic (DZ) males. Females and males have similar rates and these are negligible before age 60 for both MZ and DZ pairs. We moreover find that having a co-twin surviving to old ages substantially and significantly increases the chance of reaching the same old age and this chance is higher for MZ than for DZ twins. The relative recurrence risk of reaching age 92 is 4.8 (2.2, 7.5) for MZ males, which is significantly greater than the 1.8 (0.10, 3.4) for DZ males. The patterns for females and males are very similar, but with a shift of the female pattern with age that corresponds to the better female survival. Similar results arise when considering only those Nordic twins that survived past 75 years of age. The present large population based study shows genetic influence on human lifespan. While the estimated overall strength of genetic influence is compatible with previous studies, we find that genetic influences on lifespan are minimal prior to age 60 but increase thereafter. These findings provide a support for the search for genes affecting longevity in humans, especially at advanced ages.
Link
February 04, 2006
What is your voice pitch?
Continuing the previous post, finally you have a way of figuring out what your voice pitch is, and to see how you measure up compared with other males.
According to the above cited article, the testees' recordings had:
mean length=17.9 s, mean F0=113.2 Hz, F0 range=85.6-154.6 Hz
F0 is a correlate of pitch and is the parameter that you must estimate.
According to the authors, testees had to read an excerpt from the Rainbow Passage:
I have no idea which excerpt they used, so I said the whole thing, which took me around 90 seconds, as opposed to the 17.9 average recording length in their experiments.
Ok, so how do you do it?
First download the Praat software which is available for different platforms and then run it.
Then do "New->Record Mono Sound" and speak the passage into your microphone. Then do "Save to list".
Now, select the recording and click edit. In the new window that will appear, go to Pitch->Pitch Settings and set pitch range from 75 to 300Hz.
Then, select the part of your recording that you contains your speech by clicking at the beginning and dragging the mouse to the end.
Then View->Show Analyses and make the "Longest Analysis" as long as your recording.
Finally, Pitch->Get Pitch, and there you have it!
My own pitch is 119Hz over the entire passage. I did notice that as I was speaking and getting more impatient my voice started to rise in tone. I then calculated the pitch in the first half of the recording (117Hz) vs. the second half (121Hz) and indeed it seemed that this was the case. In the first 17.9 seconds of the recording, which corresponds to the mean length of the authors' recordings, my pitch was 116Hz.
Leave comment or trackback if you manage to get through the process; it's easier than might seem from the above description.
Update
David Puts, first author of this study writes in the comments:
According to the above cited article, the testees' recordings had:
mean length=17.9 s, mean F0=113.2 Hz, F0 range=85.6-154.6 Hz
F0 is a correlate of pitch and is the parameter that you must estimate.
According to the authors, testees had to read an excerpt from the Rainbow Passage:
When the sunlight strikes raindrops in the air, they act as a prism and form a rainbow. The rainbow is a division of white light into many beautiful colors. These take the shape of a long round arch, with its path high above, and its two ends apparently beyond the horizon. There is , according to legend, a boiling pot of gold at one end. People look, but no one ever finds it. When a man looks for something beyond his reach, his friends say he is looking for the pot of gold at the end of the rainbow. Throughout the centuries people have explained the rainbow in various ways. Some have accepted it as a miracle without physical explanation. To the Hebrews it was a token that there would be no more universal floods. The Greeks used to imagine that it was a sign from the gods to foretell war or heavy rain. The Norsemen considered the rainbow as a bridge over which the gods passed from earth to their home in the sky. Others have tried to explain the phenomenon physically. Aristotle thought that the rainbow was caused by reflection of the sun's rays by the rain. Since then physicists have found that it is not reflection, but refraction by the raindrops which causes the rainbows. Many complicated ideas about the rainbow have been formed. The difference in the rainbow depends considerably upon the size of the drops, and the width of the colored band increases as the size of the drops increases. The actual primary rainbow observed is said to be the effect of super-imposition of a number of bows. If the red of the second bow falls upon the green of the first, the result is to give a bow with an abnormally wide yellow band, since red and green light when mixed form yellow. This is a very common type of bow, one showing mainly red and yellow, with little or no green or blue.
I have no idea which excerpt they used, so I said the whole thing, which took me around 90 seconds, as opposed to the 17.9 average recording length in their experiments.
Ok, so how do you do it?
First download the Praat software which is available for different platforms and then run it.
Then do "New->Record Mono Sound" and speak the passage into your microphone. Then do "Save to list".
Now, select the recording and click edit. In the new window that will appear, go to Pitch->Pitch Settings and set pitch range from 75 to 300Hz.
Then, select the part of your recording that you contains your speech by clicking at the beginning and dragging the mouse to the end.
Then View->Show Analyses and make the "Longest Analysis" as long as your recording.
Finally, Pitch->Get Pitch, and there you have it!
My own pitch is 119Hz over the entire passage. I did notice that as I was speaking and getting more impatient my voice started to rise in tone. I then calculated the pitch in the first half of the recording (117Hz) vs. the second half (121Hz) and indeed it seemed that this was the case. In the first 17.9 seconds of the recording, which corresponds to the mean length of the authors' recordings, my pitch was 116Hz.
Leave comment or trackback if you manage to get through the process; it's easier than might seem from the above description.
Update
David Puts, first author of this study writes in the comments:
In response, to Jason Malloy's question, the male participants were racially mixed--mostly white, but maybe 10 (out of 111) black. I didn't find a significant difference in F0 between ethnicities.
Regarding Gaius' question, yes, Dabbs and Mallinger (1999) (cited in the paper) found a significant correlation between circulating testosterone levels and F0 in males of this age group. And Need et al. (1993) (also cited in our paper) found that testosterone treatment lowers voice pitch.
Finally, I think we sound kind of whiny on voicemail recordings, etc. because the microphones on these devices are not the best quality and don't pick up the entire sound spectrum very well.
Thanks for your interest!
February 03, 2006
More positive selection in recent humans: CASP12
It seems that positive selection studies are coming out in droves; you can use the search function ("positive selection") to find a bunch of them reported on this blog.
The newest study appears as a preprint in the American Journal of Human Genetics and is about a gene called human caspase-12 (CASP12) which has two variants: the active one is the original form, but the inactive one seems to have undergone positive selection beginning 60-100ky ago.
It is suggested that the inactive form confers some advantage against sepsis, which was also implicated in a frequent mtDNA haplogroup recently.
The distribution of the two versions of the gene...

... shows that the active version is largely confined to Sub-Saharan Africa. This parallels the situation in Microcephalin and ASPM.
The highest frequencies of the active version are in the Mbuti (0.60) and the San (0.57). These populations have a high frequency of non-M168 related Y-chromosomes and non-L3 related mtDNA and are thus descended from the most ancient African populations (which I have called "Paleoafricans"), rather than the more recent emergence of an population in east Africa ("Afrasians") which spawned the Eurasians and assimilated most Paleoafricans during its spread within Africa. In this regard, it is interesting that an east African population from Kenya shows the minimum (0.04) frequency of the active gene of CASP12.
American Journal of Human Genetics (in press)
Spread of an inactive form of caspase-12 in humans due to recent
positive selection
Yali Xue et al.
Abstract
The human caspase-12 gene is polymorphic for the presence or absence of a stop codon, resulting in the occurrence of both active (ancestral) and inactive (derived) forms of the gene in the population. It has previously been shown that carriers of the inactive gene are more resistant to severe sepsis. We have now investigated whether the inactive form has spread because of neutral drift or positive selection. We determined its distribution in a worldwide sample of 52 populations and re-sequenced the gene in 77 individuals from the HapMap Yoruba, Han Chinese and European populations. There is strong evidence for positive selection from low diversity, skewed allele frequency spectra and the predominance of a single haplotype. We suggest that the inactive form of the gene arose in Africa ~100-500 thousand years ago (KYA) and was initially neutral or almost neutral, but that positive selection beginning ~60-100 KYA drove it to near-fixation. We further propose that its selective advantage was sepsis resistance in populations that experienced more infectious diseases as population sizes and densities increased.
Link (pdf)
Alan Templeton vs. Out of Africa with replacement: 1-0
 Alan Templeton has followed up his work in Out of Africa again and again where he argued that the human genome holds evidence that is incompatible with an exclusively recent origin of modern humans. While human mtDNA and Y-chromosomes coalesce to a time less than 200ky and are thus contemporaneous with the emergence of anatomically modern humans, we also possess genes that are far older, and are best explained by the retention of ancestry from populations other than those emerging recently in the African continent.
Alan Templeton has followed up his work in Out of Africa again and again where he argued that the human genome holds evidence that is incompatible with an exclusively recent origin of modern humans. While human mtDNA and Y-chromosomes coalesce to a time less than 200ky and are thus contemporaneous with the emergence of anatomically modern humans, we also possess genes that are far older, and are best explained by the retention of ancestry from populations other than those emerging recently in the African continent.Templeton used classical hypothesis testing to formally reject the Out of Africa with replacement model with a staggeringly low probability. It's possible that his procedure might be contested, but I just can't see how anyone can really support the replacement theory any more.
Templeton also believes that there were several Out of Africa movements, each of them leaving a "signature" in the genomic ancestry of modern humans. Different genes are dated to each of these three movements:

Templeton also finds that the three Out of Africa movements correlate quite well with archaeologically/anthropologically established movements:
These dates overlay well upon the fossil and archaeological record, with the first expansion corresponding to the original expansion of Homo erectus out of Africa into Eurasia (Aguirre and Carbonell, 2001; Bar-Yosef and Belfer-Cohen, 2001; Anto´n et al., 2002; Vekua et al., 2002) and the development of a culture capable of keeping impaired individuals alive for many years (Lordkipanidze et al., 2005). The second expansion corresponds to the spread of Acheulean culture into much of Eurasia after an earlier African origin (Asfaw et al., 1992; Hou et al., 2000) and the initiation of a substantial increase in cranial capacity (Ruff et al., 1997; Relethford, 2001b; Rightmire, 2004). The most recent expansion out of Africa corresponds to the spread of several anatomically modern traits into Eurasia after an earlier African origin (Stringer, 2002; White et al., 2003).
This certainly also seems to agree with Erik Trinkaus' recent statement:
Versions of the assimilation model have remained contenders for the interpretation of modern human phylogenetic emergence, if frequently overshadowed by the more polarized regional continuity (with gene flow) and (out of Africa with) replacement scenarios. The last two interpretations are finally intellectually dead. Both are contradicted by available evidence, and it is time for the discussion to move on. Yet, despite the general acceptance of some form of the assimilation model, issues remain.
Update... but Milford Wolpoff writes in the comments (which disappear after 4 months on Haloscan):
I just don't see how Templeton's excellent review somehow supports Trinkaus' comment. ET asserts that multiregional evolution is "finally intellectually dead" whereas Templeton describes an interpretation of human evolution that is compatible with multiregional evolution, as he has noted. Trinkaus would replace multiregional evolution with his "assimilation theory" - an interpretation that does not seem to differ from multiregional evolution except by name, which perhaps explains the problem.
New analysis shows three human migrations out of Africa (excerpt):
Feb. 2, 2006 — A new, more robust analysis of recently derived human gene trees by Alan R. Templeton, Ph.D, of Washington University in St Louis, shows three distinct major waves of human migration out of Africa instead of just two, and statistically refutes — strongly — the 'Out of Africa' replacement theory.
That theory holds that populations of Homo sapiens left Africa 100,000 years ago and wiped out existing populations of humans. Templeton has shown that the African populations interbred with the Eurasian populations — thus, making love, not war.
"The 'Out of Africa' replacement theory has always been a big controversy," Templeton said. "I set up a null hypothesis and the program rejected that hypothesis using the new data with a probability level of 10 to the minus 17th. In science, you don't get any more conclusive than that. It says that the hypothesis of no interbreeding is so grossly incompatible with the data, that you can reject it."
Templeton's analysis is considered to be the only definitive statistical test to refute the theory, dominant in human evolution science for more than two decades.
"Not only does the new analysis reject the theory, it demolishes it," Templeton said.
Templeton published his results in the Yearbook of Physical Anthropology, 2005.
American Journal of Physical Anthropology
Volume 128, Issue S41 , Pages 33 - 59
Haplotype Trees and Modern Human Origins (p 33-59)
Alan R. Templeton
Abstract
A haplotype is a multisite haploid genotype at two or more polymorphic sites on the same chromosome in a defined DNA region. An evolutionary tree of the haplotypes can be estimated if the DNA region had little to no recombination. Haplotype trees can be used to reconstruct past human gene-flow patterns and historical events, but any single tree captures only a small portion of evolutionary history, and is subject to error. A fuller view of human evolution requires multiple DNA regions, and errors can be minimized by cross-validating inferences across loci. An analysis of 25 DNA regions reveals an out-of-Africa expansion event at 1.9 million years ago. Gene flow with isolation by distance was established between African and Eurasian populations by about 1.5 million years ago, with no detectable interruptions since. A second out-of-Africa expansion occurred about 700,000 years ago, and involved interbreeding with at least some Eurasian populations. A third out-of-Africa event occurred around 100,000 years ago, and was also characterized by interbreeding, with the hypothesis of a total Eurasian replacement strongly rejected (P < 10-17). This does not preclude the possibility that some Eurasian populations could have been replaced, and the status of Neanderthals is indecisive. Demographic inferences from haplotype trees have been inconsistent, so few definitive conclusions can be made at this time. Haplotype trees from human parasites offer additional insights into human evolution and raise the possibility of an Asian isolate of humanity, but once again not in a definitive fashion. Haplotype trees can also indicate which genes were subject to positive selection in the lineage leading to modern humans. Genetics provides many insights into human evolution, but those insights need to be integrated with fossil and archaeological data to yield a fuller picture of the origin of modern humans.
Link
February 01, 2006
IQ at age 18-20 predicts mortality 30 years later
This is probably due to the fact that IQ is a signal of good overall health, and that different occupational and social worlds are inhabited by people depending on their cognitive ability.
Int J Epidemiol. 2006 Jan 30; [Epub ahead of print]
The association between cognitive ability measured at ages 18-20 and mortality during 30 years of follow-up--a prospective observational study among Swedish males born 1949-51.
Hemmingsson T, Melin B, Allebeck P, Lundberg I.
OBJECTIVES: An association between childhood cognitive ability measured with IQ tests and mortality has been reported recently. It is not clear from those studies if the relative risk is increased only among those in the lower end of the IQ score scale or if there is graded increase in mortality from the lowest to the highest. This study aims to investigate the association between cognitive ability measured at age 18-20 and mortality during a 30 year period of follow-up. METHODS: Data on cognitive ability was collected from 49 323 men, born in 1949-51, who were conscripted for compulsory military training in 1969/70. Data on mortality were obtained from the Causes of Death register 1971-2000. RESULTS: Cognitive ability was a strong predictor of all-cause mortality, cardiovascular disease (CVD)-mortality, mortality from violent causes, and alcohol-related mortality. A striking finding was a pronounced gradient in mortality risk across all IQ score groups. Adjustment for adult socioeconomic position attenuated the increased risk somewhat [for all-cause mortality: crude hazard ratio (HR) 1.16 (1.13-1.19), adjusted HR 1.12 (1.09-1.15)]. CONCLUSION: IQ test score measured in late adolescence (only males) was a significant predictor of all-cause, as well as cause-specific (CVD and injuries), mortality during 30 years of follow-up. The risk increased from high to low IQ test score results for all outcomes.
Link
Int J Epidemiol. 2006 Jan 30; [Epub ahead of print]
The association between cognitive ability measured at ages 18-20 and mortality during 30 years of follow-up--a prospective observational study among Swedish males born 1949-51.
Hemmingsson T, Melin B, Allebeck P, Lundberg I.
OBJECTIVES: An association between childhood cognitive ability measured with IQ tests and mortality has been reported recently. It is not clear from those studies if the relative risk is increased only among those in the lower end of the IQ score scale or if there is graded increase in mortality from the lowest to the highest. This study aims to investigate the association between cognitive ability measured at age 18-20 and mortality during a 30 year period of follow-up. METHODS: Data on cognitive ability was collected from 49 323 men, born in 1949-51, who were conscripted for compulsory military training in 1969/70. Data on mortality were obtained from the Causes of Death register 1971-2000. RESULTS: Cognitive ability was a strong predictor of all-cause mortality, cardiovascular disease (CVD)-mortality, mortality from violent causes, and alcohol-related mortality. A striking finding was a pronounced gradient in mortality risk across all IQ score groups. Adjustment for adult socioeconomic position attenuated the increased risk somewhat [for all-cause mortality: crude hazard ratio (HR) 1.16 (1.13-1.19), adjusted HR 1.12 (1.09-1.15)]. CONCLUSION: IQ test score measured in late adolescence (only males) was a significant predictor of all-cause, as well as cause-specific (CVD and injuries), mortality during 30 years of follow-up. The risk increased from high to low IQ test score results for all outcomes.
Link
Dancers vs. Non-Dancers: Genes and Personality
Are dancers genetically different than the rest of us? Yes, says Hebrew University researcher
PLoS Genetics Volume 1 Issue 3 SEPTEMBER 2005
AVPR1a and SLC6A4 Gene Polymorphisms Are Associated with Creative Dance Performance
Rachel Bachner-Melman et al.
Dancing, which is integrally related to music, likely has its origins close to the birth of Homo sapiens, and throughout our history, dancing has been universally practiced in all societies. We hypothesized that there are differences among individuals in aptitude, propensity, and need for dancing that may partially be based on differences in common genetic polymorphisms. Identifying such differences may lead to an understanding of the neurobiological basis of one of mankind's most universal and appealing behavioral traits—dancing. In the current study, 85 current performing dancers and their parents were genotyped for the serotonin transporter (SLC6A4: promoter region HTTLPR and intron 2 VNTR) and the arginine vasopressin receptor 1a (AVPR1a: promoter microsatellites RS1 and RS3). We also genotyped 91 competitive athletes and a group of nondancers/nonathletes (n = 872 subjects from 414 families). Dancers scored higher on the Tellegen Absorption Scale, a questionnaire that correlates positively with spirituality and altered states of consciousness, as well as the Reward Dependence factor in Cloninger's Tridimensional Personality Questionnaire, a measure of need for social contact and openness to communication. Highly significant differences in AVPR1a haplotype frequencies (RS1 and RS3), especially when conditional on both SLC6A4 polymorphisms (HTTLPR and VNTR), were observed between dancers and athletes using the UNPHASED program package (Cocaphase: likelihood ratio test [LRS] = 89.23, p = 0.000044). Similar results were obtained when dancers were compared to nondancers/nonathletes (Cocaphase: LRS = 92.76, p = 0.000024). These results were confirmed using a robust family-based test (Tdtphase: LRS = 46.64, p = 0.010). Association was also observed between Tellegen Absorption Scale scores and AVPR1a (Qtdtphase: global chi-square = 26.53, p = 0.047), SLC6A4 haplotypes (Qtdtphase: chi-square = 2.363, p = 0.018), and AVPR1a conditional on SCL6A4 (Tdtphase: LRS = 250.44, p = 0.011). Similarly, significant association was observed between Tridimensional Personality Questionnaire Reward Dependence scores and AVPR1a RS1 (chi-square = 20.16, p = 0.01). Two-locus analysis (RS1 and RS3 conditional on HTTLPR and VNTR) was highly significant (LRS = 162.95, p = 0.001). Promoter repeat regions in the AVPR1a gene have been robustly demonstrated to play a role in molding a range of social behaviors in many vertebrates and, more recently, in humans. Additionally, serotonergic neurotransmission in some human studies appears to mediate human religious and spiritual experiences. We therefore hypothesize that the association between AVPR1a and SLC6A4 reflects the social communication, courtship, and spiritual facets of the dancing phenotype rather than other aspects of this complex phenotype, such as sensorimotor integration.
Link
What makes dancers different than the rest of us? Genetic variants, says a researcher at the Hebrew University of Jerusalem.
In a study published in the American journal, Public Library of Science Genetics, Psychology Prof. Richard P. Ebstein and his research associates have shown, through DNA examination, that dancers show consistent differences in two key genes from the general population. Ebstein is the head of the Hebrew University Psychology Department's Scheinfeld Center for Human Genetics in the Social Sciences.
This finding is not surprising, says Ebstein, in view of other studies of musicians and athletes, which also have shown genetic differences.
Ebstein and his colleagues found in an examination of 85 dancers and advanced dancing students in Israel variants of two genes that provide the code for the serotonin transporter and arginine vasopressin receptor 1a.
Both genes are involved in the transmission of information between nerve cells. The serotonin transporter regulates the level of serotonin, a brain transmitter that contributes to spiritual experience, among many other behavioral traits. The vasopressin receptor has been shown in many animal studies to modulate social communication and affiliative bonding behaviors. Both are elements involved in the age-old human social expression of dancing.
The genetic evidence was corroborated by two questionnaires distributed by the researchers to the dancers. One is the Tellegen Absorption Scale (TAS), that correlates aspects of spirituality and altered states of consciousness, and the other is the Tridimensional Personality Questionnaire (TPQ), a measure of the need for social contact and openness to communication.
The genetic and questionnaire results of the dancers were compared with those of two other groups examined – athletes as well as those who were both non-dancers and non-athletes. (Athletes were chosen for comparison since they require a good deal of physical stamina like dancers.)
When the results were combined and analyzed, it was clearly shown that the dancers exhibited particular genetic and personality characteristics that were not found in the other two groups.
The dancer "type," says Ebstein, clearly demonstrates qualities that are not necessarily lacking but are not expressed as strongly in other people: a heightened sense of communication, often of a symbolic and ceremonial nature, and a strong spiritual personality trait.
Others involved in the research with Ebstein were his Ph.D. student Rachel Bachner- Melman, as well as additional researchers from Israel and France.
PLoS Genetics Volume 1 Issue 3 SEPTEMBER 2005
AVPR1a and SLC6A4 Gene Polymorphisms Are Associated with Creative Dance Performance
Rachel Bachner-Melman et al.
Dancing, which is integrally related to music, likely has its origins close to the birth of Homo sapiens, and throughout our history, dancing has been universally practiced in all societies. We hypothesized that there are differences among individuals in aptitude, propensity, and need for dancing that may partially be based on differences in common genetic polymorphisms. Identifying such differences may lead to an understanding of the neurobiological basis of one of mankind's most universal and appealing behavioral traits—dancing. In the current study, 85 current performing dancers and their parents were genotyped for the serotonin transporter (SLC6A4: promoter region HTTLPR and intron 2 VNTR) and the arginine vasopressin receptor 1a (AVPR1a: promoter microsatellites RS1 and RS3). We also genotyped 91 competitive athletes and a group of nondancers/nonathletes (n = 872 subjects from 414 families). Dancers scored higher on the Tellegen Absorption Scale, a questionnaire that correlates positively with spirituality and altered states of consciousness, as well as the Reward Dependence factor in Cloninger's Tridimensional Personality Questionnaire, a measure of need for social contact and openness to communication. Highly significant differences in AVPR1a haplotype frequencies (RS1 and RS3), especially when conditional on both SLC6A4 polymorphisms (HTTLPR and VNTR), were observed between dancers and athletes using the UNPHASED program package (Cocaphase: likelihood ratio test [LRS] = 89.23, p = 0.000044). Similar results were obtained when dancers were compared to nondancers/nonathletes (Cocaphase: LRS = 92.76, p = 0.000024). These results were confirmed using a robust family-based test (Tdtphase: LRS = 46.64, p = 0.010). Association was also observed between Tellegen Absorption Scale scores and AVPR1a (Qtdtphase: global chi-square = 26.53, p = 0.047), SLC6A4 haplotypes (Qtdtphase: chi-square = 2.363, p = 0.018), and AVPR1a conditional on SCL6A4 (Tdtphase: LRS = 250.44, p = 0.011). Similarly, significant association was observed between Tridimensional Personality Questionnaire Reward Dependence scores and AVPR1a RS1 (chi-square = 20.16, p = 0.01). Two-locus analysis (RS1 and RS3 conditional on HTTLPR and VNTR) was highly significant (LRS = 162.95, p = 0.001). Promoter repeat regions in the AVPR1a gene have been robustly demonstrated to play a role in molding a range of social behaviors in many vertebrates and, more recently, in humans. Additionally, serotonergic neurotransmission in some human studies appears to mediate human religious and spiritual experiences. We therefore hypothesize that the association between AVPR1a and SLC6A4 reflects the social communication, courtship, and spiritual facets of the dancing phenotype rather than other aspects of this complex phenotype, such as sensorimotor integration.
Link
Subscribe to:
Posts (Atom)