Ancient DNA has consistently managed to surprise us, with pretty much no direct genetic continuity revealed between Pleistocene and modern populations anywhere in the world. So, it is refreshing to see that at least in the case of the Americas the people who lived there ~13 thousand years ago are clearly related to the people who lived there in pre-Columbian times, with no real evidence of subsequent gene flows from Eurasia (at least in the case of Central/South Americans).
Many people suspected this because of the difficulty to access the Americas from Eurasia: this must have limited gene flow between the two regions to a handful of migrants and a restricted set of time periods where geological and climatic conditions were advantageous. The much reduced genetic diversity of Native Americans also argues in favor of them being a relatively simple population, with low heterozygosity and a handful of unique "founder lineages" in both the Y-chromosome and mtDNA.
Nonetheless, there are also several theories in the realm of alernative history, involving Solutreans from Europe, trans-Pacific boat riders, bearded "White Gods", Minoans/Phoenicians/Atlanteans/Ancient Egyptians, "African" Olmecs, "Caucasoid" Paleo-Indians, lost Israelite tribes, to mention only a few of the most well-known ones.
The new study does not, of course, disprove any of the proposals in the preceding paragraph: one can still claim that diverse groups once inhabited the Americas and Rasmussen et al. (2014) just happened to chance upon one that looked just like modern native Americans. But, this certainly improves the odds of early "Native American simplicity", offering no evidence for the complexity postulated by many of the alternative theories.
Moreover, while the existence of other human groups in the Americas cannot be disproved by the study of a single ancient individual, what
can be proved is the antiquity of the ancestors of Native Americans. Rather than being late arrivals arriving from Asia after the initial colonization, perhaps with derived Mongoloid physical morphology, we now know that they were already there as early as ~13 thousand years ago. It is remarkable that a single ancient DNA sample can sweep away much of the nonsense that has been written on the topic in the past.
A piece in
Nature News addresses some of the "ethics" debate that seems ever-present in studies involving Native American remains. I don't know how this study will be perceived by living Native Americans: a possibility is that they'll be more receptive to ancient DNA research now that a team of scientists have stretched the time depth of their ancestry in the Americas to the earliest studied sample, revealing themselves not to be the evil-doers that western scientists are generally assumed to be according to a certain kind of mentality. A different -and more alarming- possibility, is that radical anti-science elements will be emboldened by these findings to claim that continuity with the earliest Americans (which in itself seems true enough) adds support to claims of ownership to pretty much all archaeological samples whose relationship to living Amerindians was hitherto uncertain in light of the many alternative theories.
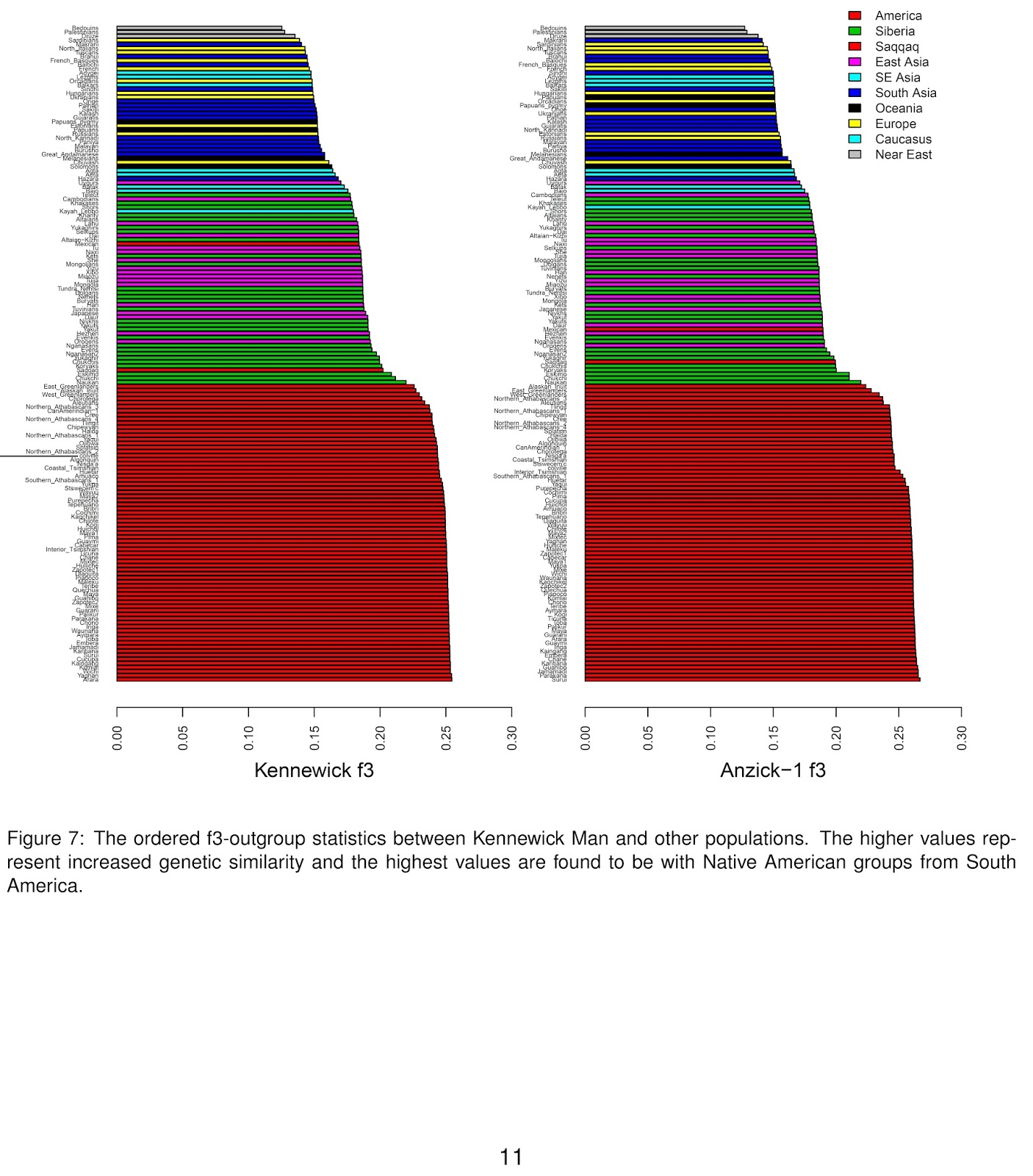
In any case, it is remarkable that this ~13 thousand year old genome now exists while the genomes of modern native Americans that can be had for a fraction of the cost and technical difficulty do not. Indeed, not even genotype data exist from most Amerindian groups from the USA, which creates the rather bizarre state of affairs that the Anzick-1 genome had to be compared with native groups from several countries in the Western hemisphere except the one in which it was found.
Nature 506, 225–229 (13 February 2014) doi:10.1038/nature13025
The genome of a Late Pleistocene human from a Clovis burial site in western Montana
Morten Rasmussen et al.
Clovis, with its distinctive biface, blade and osseous technologies, is the oldest widespread archaeological complex defined in North America, dating from 11,100 to 10,700 14C years before present (BP) (13,000 to 12,600 calendar years BP)1, 2. Nearly 50 years of archaeological research point to the Clovis complex as having developed south of the North American ice sheets from an ancestral technology3. However, both the origins and the genetic legacy of the people who manufactured Clovis tools remain under debate. It is generally believed that these people ultimately derived from Asia and were directly related to contemporary Native Americans2. An alternative, Solutrean, hypothesis posits that the Clovis predecessors emigrated from southwestern Europe during the Last Glacial Maximum4. Here we report the genome sequence of a male infant (Anzick-1) recovered from the Anzick burial site in western Montana. The human bones date to 10,705 ± 35 14C years BP (approximately 12,707–12,556 calendar years BP) and were directly associated with Clovis tools. We sequenced the genome to an average depth of 14.4× and show that the gene flow from the Siberian Upper Palaeolithic Mal’ta population5 into Native American ancestors is also shared by the Anzick-1 individual and thus happened before 12,600 years BP. We also show that the Anzick-1 individual is more closely related to all indigenous American populations than to any other group. Our data are compatible with the hypothesis that Anzick-1 belonged to a population directly ancestral to many contemporary Native Americans. Finally, we find evidence of a deep divergence in Native American populations that predates the Anzick-1 individual.
Link