Not really anthropological-related, but I was looking for a way to switch the comments on the blog from Haloscan to Blogger and not lose all the existing past comments. As far as I could tell there is no "clean" way to achieve this, so this is my work-around.
First of all, if you just replaced the haloscan code in your template with the blogger code, this would result in all your previous posts having new blogger comments. So, that solution is out.
I simply created a new user account for posting in my blog, whose blogger profile name is "dienekesp" rather than "Dienekes". Now, when I post as "dienekesp", the <$BlogItemAuthor$> tag takes the value "dienekesp".
Next, you place both the haloscan comment-generating code, and the blogger comment-generating code in your template, and you "comment out" the code you don't want to use by two simple JavaScripts:
<script type="text/javascript"><!--
if ("<$BlogItemAuthor$>"!="Dienekes")
document.write("\< !--"); //-->
</script>
BLOGGER COMMENT CODE
<script type="text/javascript"><!--
if ("<$BlogItemAuthor$>"!="Dienekes")
document.write("--\>");
//-->;
</script>
The same code as above should also be repeated for the Haloscan comments code, but with "dienekesp" instead of "Dienekes". So, from now on whenever I post as "dienekesp", Blogger comments will be used; when I post as "Dienekes", Haloscan comments will be used. All the older posts which had been posted as "Dienekes" will retain their existing comments.
PS: It goes without saying that this trick is provided as-is with no guarantee that it will work or acceptance of responsibility if it doesn't or you wreck your blog in your process. Make sure you back up your template in advance.

February 29, 2008
Did Cannibalism contribute to Neanderthal extinction?
See the earlier post on Cannibalistic Neanderthals.
Medical Hypotheses doi:10.1016/j.mehy.2007.12.014
A potential role for Transmissible Spongiform Encephalopathies in Neanderthal extinction
Simon Underdown
The Neanderthals were a Eurasian human species of the genus Homo that disappeared approximately 30,000 years ago. The cause or causes of their extinction continues to intrigue specialists and non-specialists alike. Here a contributory role for Transmissible Spongiform Encephalopathies (TSEs) is suggested. TSEs could have infected Neanderthal groups as a result of general cannibalistic activity and brain tissue consumption in particular. Further infection could then have taken place through continued cannibalistic activity or via shared used of infected stone tools. A modern human hunter-gatherer proxy has been developed and applied as a hypothetical model to the Neanderthals. This hypothesis suggests that the impact of TSEs on the Neanderthals could have been dramatic and have played a large part in contributing to the processes of Neanderthal extinction.
Link
Medical Hypotheses doi:10.1016/j.mehy.2007.12.014
A potential role for Transmissible Spongiform Encephalopathies in Neanderthal extinction
Simon Underdown
The Neanderthals were a Eurasian human species of the genus Homo that disappeared approximately 30,000 years ago. The cause or causes of their extinction continues to intrigue specialists and non-specialists alike. Here a contributory role for Transmissible Spongiform Encephalopathies (TSEs) is suggested. TSEs could have infected Neanderthal groups as a result of general cannibalistic activity and brain tissue consumption in particular. Further infection could then have taken place through continued cannibalistic activity or via shared used of infected stone tools. A modern human hunter-gatherer proxy has been developed and applied as a hypothetical model to the Neanderthals. This hypothesis suggests that the impact of TSEs on the Neanderthals could have been dramatic and have played a large part in contributing to the processes of Neanderthal extinction.
Link
East-West cranial differentiation in Central and Northern America
 A very related previous blog entry on East-west cranial differences of South Americans. Also read a major study on Native American genetic variation. Also of interest, the Peopling of the Americas, and New synthesis on the first arrivals into the New World. Directly related to the issue of East-West differentiation in the Americas is the post on South American mtDNA, re-interpretation of its prehistory.
A very related previous blog entry on East-west cranial differences of South Americans. Also read a major study on Native American genetic variation. Also of interest, the Peopling of the Americas, and New synthesis on the first arrivals into the New World. Directly related to the issue of East-West differentiation in the Americas is the post on South American mtDNA, re-interpretation of its prehistory.Journal of Human Evolution Volume 54, Issue 3, March 2008, Pages 296-308
East-West cranial differentiation in pre-Columbian populations from Central and North America
Héctor M. Pucciarelli
Abstract
In a recent study we found that crania from South Amerindian populations on each side of the Andes differ significantly in terms of craniofacial shape. Western populations formed one morphological group, distributed continuously over 14,000 km from the Fuegian archipelago (southern Chile) to the Zulia region (northwestern Venezuela). Easterners formed another group, distributed from the Atlantic Coast up to the eastern foothills of the Andes. This differentiation is further supported by several genetic studies, and indirectly by ecological and archaeological studies. Some authors suggest that this dual biological pattern is consistent with differential rates of gene flow and genetic drift operating on both sides of the Cordillera due to historical reasons. Here we show that such East-West patterning is also observable in North America. We suggest that the “ecological zones model” proposed by Dixon, explaining the spread of the early Americans along a Pacific dispersal corridor, combined with the evolution of different population dynamics in both regions, is the most parsimonious mechanism to explain the observed patterns of within- and between-group craniofacial variability.
Link
Domesticated cattle of predominantly Neolithic origin but with some aurochs mtDNA
 See also The origin of European cattle, and Neolithic Near Eastern Origin of domesticated cattle. Some introductory information about the aurochs via the BBC and Wikipedia and the Extinction Website.
See also The origin of European cattle, and Neolithic Near Eastern Origin of domesticated cattle. Some introductory information about the aurochs via the BBC and Wikipedia and the Extinction Website.Curr Biol. 2008 Feb 26;18(4):R157-8.
Mitochondrial genomes of extinct aurochs survive in domestic cattle.
Achilli A, Olivieri A, Pellecchia M, Uboldi C, Colli L, Al-Zahery N, Accetturo M, Pala M, Kashani BH, Perego UA, Battaglia V, Fornarino S, Kalamati J, Houshmand M, Negrini R, Semino O, Richards M, Macaulay V, Ferretti L, Bandelt HJ, Ajmone-Marsan P, Torroni A.
Archaeological and genetic evidence suggest that modern cattle might result from two domestication events of aurochs (Bos primigenius) in southwest Asia, which gave rise to taurine (Bos taurus) and zebuine (Bos indicus) cattle, respectively [1-3]. However, independent domestication in Africa [4,5] and East Asia [6] has also been postulated and ancient DNA data raise the possibility of local introgression from wild aurochs [7-9]. Here, we show by sequencing entire mitochondrial genomes from modern cattle that extinct wild aurochsen from Europe occasionally transmitted their mitochondrial DNA (mtDNA) to domesticated taurine breeds. However, the vast majority of mtDNAs belong either to haplogroup I (B. indicus) or T (B. taurus). The sequence divergence within haplogroup T is extremely low (eight-fold less than in the human mtDNA phylogeny [10]), indicating a narrow bottleneck in the recent evolutionary history of B. taurus. MtDNAs of haplotype T fall into subclades whose ages support a single Neolithic domestication event for B. taurus in the Near East, 9-11 thousand years ago (kya).
Link (pdf)
February 28, 2008
Migrations in the Baltic region inferred from Y chromosomes and mtDNA

From the paper on Y-haplogroup I1a:
Haplogroup I1a is suggested to have its origins in the Iberian refugium, from where it spread northward and now has its highest frequencies in Northern Europe (Rootsi et al. 2004). The haplotype matches to Germany and Poland imply that I1a has arrived to the Nordic countries from the Southern Baltic Sea region, which is historically plausible. The coalescense age of the haplogroup is about 5000 years lower than the age of the earliest archaeological findings from the Northern Baltic Sea region, which suggests a Neolithic arrival. There are two possible migration routes from Central Europe to the Northern Baltic Sea region: an exclusive western route via Sweden, an eastern route via the Baltic states, or via both to Eastern Finland and Karelia (Fig. 5). The surprisingly high diversities of I1a among the eastern Finnish and Baltic populations, and the lack of association between the Western Finns and the Swedes in SAMOVA analysis suggest that I1a has been involved in bifurcating migrations both via Sweden and the Baltic states, and that the presence of the haplogroup in Finland and Karelia is not merely due to Swedish influence. The low frequency of I1a among the Baltic populations may be due to later effects of genetic drift or replacement.
I am personally doubtful of the Iberian origin of Y-haplogroup I1a. Its presence in Iberia and France, as well as its high diversity there may be the result of migration from northern Europe, a genetic trace of the Germanic Volkerwanderung. One certainly needs to consider this effect. There are probably a couple of papers just waiting to be written by looking at Y-chromosomes of Germanic descendants in Southwestern Europe by looking at either surnames or early cemetaries.
With regard to Y-haplogroup N3:
The frequency distribution and age of haplogroup N3 in our study sample was consistent with the earlier studies (Lahermo et al. 1999, Zerjal et al. 2001, Tambets et al. 2004, Karlsson et al. 2006, Rootsi et al. 2007). According to the YHRD database, the haplotypes most common in Finland and Karelia were relatively unique, which is not unexpected, since data from most Eurasian populations where N3 is common is not publicly available. It seems evident that the Finns and Karelians share a history regarding haplogroup N3. In the database comparisons, we also observed that N3 may mark a westward diffusion in the north from Finland to Sweden and in the south from the Baltic countries to Poland and Germany.The researchers also reiterated the previous idea of a dual origin of N3, as shown in the figure, with different clades being represented in Finland and the Baltic states, with Estonia being intermediate between the two.
With regard to Y-haplogroup R1a1:
It is plausible that both R1a1 and I1a were carried to the Baltic Sea region via the same Neolithic migrations from Germany/Poland. The higher coalescence age and the starlike network structure of R1a1 are consistent with the probable higher diversity and frequency of R1a1 in the original source population(s), a consequence of the wider geographical distribution of the haplogroup. It is an important observation that in the Baltic Sea region R1a1 is mainly associated to Central European rather than eastern or Russian influence. However, haplotype frequency comparisons (Derenko et al. 2006, Willuweit & Roewer 2007) give some indication of Russian gene flow as a partial source of R1a1 in Karelia, which would be plausible given the long period of admixture with Slavs (Fig. 5). However, the Y-chromosomal diversity in Karelia has been heavily affected by drift and founder effects. Another haplogroup with eastern affinity is I1b (Rootsi et al. 2004), whose presence in Karelia and the Baltic states is probably a sign of Russian gene flow.
This is an important discovery, and a great first step in uncovering the structure within this widespread haplogroup. Even though R1a1 Y-chromosomes were studied in a lot of populations, including e.g., the Balkans, India, the Altai, unfortunately we know next to nothing about its phylogenetic substructure. Without such knowledge, R1a1 spread has been variously interpreted as a signal of postglacial colonization, Kurgan expansions, or the spread of Slavic languages.
Finally, the genetic legacy of the Saami is visible in mtDNA:
The eastern elements in the mtDNA variation of the Baltic Sea region are intertwined with the Saami influence. Recent studies of the mtDNA variation among the Saami show a link to the Volga-Ural region (Tambets et al. 2004, Ingman & Gyllensten 2006), which is now shown to exist also among the Karelians and, to a lesser degree, among the other populations from the Baltic Sea region as well. Additionally, the presence of U4 in the Eastern Baltic Sea populations may represent eastern influence, since it is typical for the Volga-Ural region (Bermisheva et al. 2002). The high diversity of this haplogroup in the Baltic region, observable in the haplotype network, suggests a complex history, and rules genetic drift out as a cause of the high frequency. All in all, these mtDNA haplogroups may be maternal reflections of the eastern influence that can be most clearly observed in the Y-chromosomal haplogroup N3.
Annals of Human Genetics doi:10.1111/j.1469-1809.2007.00429.x
Migration Waves to the Baltic Sea Region
T. Lappalainen et al.
In this study, the population history of the Baltic Sea region, known to be affected by a variety of migrations and genetic barriers, was analyzed using both mitochondrial DNA and Y-chromosomal data. Over 1200 samples from Finland, Sweden, Karelia, Estonia, Setoland, Latvia and Lithuania were genotyped for 18 Y-chromosomal biallelic polymorphisms and 9 STRs, in addition to analyzing 17 coding region polymorphisms and the HVS1 region from the mtDNA. It was shown that the populations surrounding the Baltic Sea are genetically similar, which suggests that it has been an important route not only for cultural transmission but also for population migration. However, many of the migrations affecting the area from Central Europe, the Volga-Ural region and from Slavic populations have had a quantitatively different impact on the populations, and, furthermore, the effects of genetic drift have increased the differences between populations especially in the north. The possible explanations for the high frequencies of several haplogroups with an origin in the Iberian refugia (H1, U5b, I1a) are also discussed.
Link
February 25, 2008
The Macedonian Issue

The issue of the naming dispute between Greece and the Former Yugoslav Republic of Macedonia (FYROM) is currently in the news, so this is a good time to address some facts pertaining to it.
The dispute centers around the issue of the use of the adjective "Macedonian". This adjective has a geographical sense, describing someone who is from the geographical region of Macedonia. However, for the inhabitants of FYROM, it also has an ethnic sense, since many (or most) Slavic-speaking inhabitants of FYROM consider themselves to be Macedonian in an ethnic sense. I will distinguish between these two uses as Geo-Macedonians and Slavo-Macedonians henceforth.
Three types of criteria are used with regard to ethnic identification: language, self-definition, descent.
Descent is the easier one to address: according to most reputable genetic studies, Geo-Macedonians tend to be more of indigenous Balkan origins. In short, Geo-Macedonians, whether they speak Greek, Vlach, Albanian, or Slavic, and whether they live in Greece or the FYROM are characterized by genetic characteristics attesting to a substantial amount of distinctive southern Balkan ancestry, having, in particular substantial frequencies of Y-haplogroups E and J.
Genetically-speaking Geo-Macedonians are Balkan Europeans, and are differentiated both from Northern-Balkan/Central/Eastern Europeans as well as from West Asians.
Language is also easy to address. Within Geo-Macedonia, several languages and dialects are spoken, including Greek, Slavic, Vlach, and Albanian. These languages were established at different times, with the oldest attested one being the Greek. A Slavic language spoken in the FYROM is named "Macedonian" by the Slavo-Macedonians.
Sometimes, it is asserted by scholars from FYROM that Ancient Macedonian was not a Greek dialect, but a separate Indo-European language. While these claims are not very serious, they are actually irrelevant to the modern issues at hand, since the "Macedonian" of the Slavo-Macedonians has no relationship to the ancient language: it is a Slavic idiom dating at most from the 6th c. AD.
Self-Identity is more complex. It is true that Slavic speakers from FYROM have a Macedonian ethnic identity, but Geo-Macedonians in Greece have a Greek ethnic identity. Thus, Greek Macedonians assert the name "Macedonian" in a geographical sense, i.e., "Greeks from the region of Macedonia", whereas Slavo-Macedonians assert it in an ethnic sense, i.e., "Members of the Macedonian ethnic group".
To summarize, we must make these points:
- Slavo-Macedonians from FYROM assert a Macedonian ethnicity, as well as (in some cases) continuity with ancient Macedonia
- Such continuity with ancient Macedonia can be based either on language, or on descent.
- Linguistic continuity between Slavic Macedonian and the ancient Macedonian dialect/language does not exist.
- Continuity of descent from the ancient Macedonians to the modern ones is not limited to the Slavic-speaking Slavo-Macedonians. Slav Macedonians have no monopoly on Macedonian "blood".
- It does not encompass the entirety of Macedonia (as a geographical region).
- The Macedonian dialect/language of Slavic is not related to the ancient Macedonian language/dialect.
- The people of FYROM have a Macedonian ethnic self-identity, but the Greeks of Macedonia have a Greek ethnic self-identity. One's "right" to the name of Macedonian infringes on the other's right to use it in a different sense; the name "Macedonian" is thus ambiguous.
Republic of Upper Macedonia describes the state geographically, since FYROM includes the northern part of historical Macedonia, as well as other historically non-Macedonian regions.
Republic of Slav Macedonia describes the state linguistically, since FYROM includes the mainly Slav-speaking portion of historical Macedonia.
The name New Macedonia might also be considered, but does not really describe the identity of FYROM as clearly as the above two.
My personal preference would be for Upper Macedonia, a clear, unambiguous geographical description that has certain advantages over Slav Macedonia:
- There are Slav speakers also in Bulgarian and Greek Macedonia.
- FYROM includes also Albanian, Vlach, and Greek speakers who might not be as open to the name "Slav Macedonia".
- The people of Greece are justified in wanting to deny exclusive rights to the Macedonian name to FYROM, because FYROM encompasses only part of Macedonia: geographically, the northern part; genetically, a subset of the Macedonian blood; linguistically, a Slavic dialect of the Macedonian region.
- The people of FYROM are justified in wanting to have some rights to the name Macedonian: they inhabit parts of Macedonia, they speak a Macedonian dialect of the Slavic group, and they have come to think of themselves as a separate nation from other Balkan Slavs.
February 23, 2008
Huge paper on human genetic relationships based on 650K SNPs
If you thought that this week's Nature paper on human variation was nice, another new paper in Science will be another pleasant surprise. It seems that every time I turn my head geneticists are raising the number of SNPs they study. Lots of populations, 650K SNPs, and a treasure trove of new insight into where humans come from and how we differ from each other.
Before I get into the details of the paper, I want to reiterate my conviction that the problem of human origins is not really a hard one. It just requires a lot of data, a lot of populations, individuals sampled, a lot of genetic markers. Our history, our race, and now it seems even our ethnicity can be read off our genes. We just need to invest the money and effort to find out. With that said, it is sad that the same-old roster of populations from the CEPH panel makes yet another appearance.
There is really a lot on the paper that might interest you, but I will note a few points. First, look at the following PC plot from the paper.

No, your eyes aren't deceiving you. This paper is proof positive that European ethnicities can be distinguished from each other genetically. Even close-by populations (in this case the French and the Italians) are neatly separated. When geographic distance increases there isn't even a hint of confusion: e.g., Russians, Orcadians, and Basque are neatly and clearly separated from other groups. Doubtlessly there would be some more overlap if more individuals/population were used, but the thrust of the discovery is intact: it seems that several European ethnicities and local populations make sense not only culturally but also biologically.
Now, take a look at the standard STRUCTURE analysis which provides meaningful results up to K=7. The standard Sub-Saharan, Native American, East Asian, and Oceanian clusters are there, but now there is meaningful structure within the Caucasoids as well; they are broken into "Middle Eastern", "European", and "Central South Asian" groups.

I would guess that the "brown" Middle-Eastern cluster is largely an Arab/Semitic phenomenon, although the inclusion of the Berber Mozabites is interesting. If it reflected a pre-historic phenomenon, then it would be difficult to explain its apparent total lack of influence in Europe, except for a barely perceptible spillage into Tuscany, which once again reiterates the idiosyncratic "Middle Eastern" trace of that Italian population of likely Etruscan descendants.
The "Central South Asian" group is also extremely interesting, for several reasons. First, it reinforces the previous claim that the Kalash, rather than Greek descendants, as some romantics would have them, are simply a non-European native population, with no evidence of European ancestry. Second, it shows that there is minimal, yet evident European influence in Central Asia, which I would relate to the eastern Indo-Iranians. Third, Central Asian influence in Europe is non-evident, except for a trace among the Russians (and substantially more among the Adygei a people of the Caucasus). Fourth the minority Mongoloid and "Boreal" (Purple) influence in Russians is affirmed. Note that we are dealing with ethnic Russians from the north (Vologda oblast), and Russians are a heterogeneous people in terms of their origin.
We can only wish for inclusion of further populations in future studies of the kind. In particular, eastern and southeastern Europe and non-Arab West Asia, Siberia would be invaluable in further understanding Caucasoid origins, and perhaps uncovering additional structure.
Note that the CEPH panel has revealed that ethnic groups can be distinguished genetically, there is little more than it can offer in terms of understanding origins. The next milestone would be either to include previously unsampled populations, or to dig into ethnic groups themselves, and see if sub-ethnic entities are also discernible in our genomes. Genetic genealogists are in for a good time in the coming years...
Science 22 February 2008: Vol. 319. no. 5866, pp. 1100 - 1104
Worldwide Human Relationships Inferred from Genome-Wide Patterns of Variation
Jun Z. Li,1,2*{dagger} Devin M. Absher,1,2* Hua Tang,1 Audrey M. Southwick,1,2 Amanda M. Casto,1 Sohini Ramachandran,4 Howard M. Cann,5 Gregory S. Barsh,1,3 Marcus Feldman,4{ddagger} Luigi L. Cavalli-Sforza,1{ddagger} Richard M. Myers1,2{ddagger}
Human genetic diversity is shaped by both demographic and biological factors and has fundamental implications for understanding the genetic basis of diseases. We studied 938 unrelated individuals from 51 populations of the Human Genome Diversity Panel at 650,000 common single-nucleotide polymorphism loci. Individual ancestry and population substructure were detectable with very high resolution. The relationship between haplotype heterozygosity and geography was consistent with the hypothesis of a serial founder effect with a single origin in sub-Saharan Africa. In addition, we observed a pattern of ancestral allele frequency distributions that reflects variation in population dynamics among geographic regions. This data set allows the most comprehensive characterization to date of human genetic variation.
Link
Before I get into the details of the paper, I want to reiterate my conviction that the problem of human origins is not really a hard one. It just requires a lot of data, a lot of populations, individuals sampled, a lot of genetic markers. Our history, our race, and now it seems even our ethnicity can be read off our genes. We just need to invest the money and effort to find out. With that said, it is sad that the same-old roster of populations from the CEPH panel makes yet another appearance.
There is really a lot on the paper that might interest you, but I will note a few points. First, look at the following PC plot from the paper.

No, your eyes aren't deceiving you. This paper is proof positive that European ethnicities can be distinguished from each other genetically. Even close-by populations (in this case the French and the Italians) are neatly separated. When geographic distance increases there isn't even a hint of confusion: e.g., Russians, Orcadians, and Basque are neatly and clearly separated from other groups. Doubtlessly there would be some more overlap if more individuals/population were used, but the thrust of the discovery is intact: it seems that several European ethnicities and local populations make sense not only culturally but also biologically.
Now, take a look at the standard STRUCTURE analysis which provides meaningful results up to K=7. The standard Sub-Saharan, Native American, East Asian, and Oceanian clusters are there, but now there is meaningful structure within the Caucasoids as well; they are broken into "Middle Eastern", "European", and "Central South Asian" groups.

I would guess that the "brown" Middle-Eastern cluster is largely an Arab/Semitic phenomenon, although the inclusion of the Berber Mozabites is interesting. If it reflected a pre-historic phenomenon, then it would be difficult to explain its apparent total lack of influence in Europe, except for a barely perceptible spillage into Tuscany, which once again reiterates the idiosyncratic "Middle Eastern" trace of that Italian population of likely Etruscan descendants.
The "Central South Asian" group is also extremely interesting, for several reasons. First, it reinforces the previous claim that the Kalash, rather than Greek descendants, as some romantics would have them, are simply a non-European native population, with no evidence of European ancestry. Second, it shows that there is minimal, yet evident European influence in Central Asia, which I would relate to the eastern Indo-Iranians. Third, Central Asian influence in Europe is non-evident, except for a trace among the Russians (and substantially more among the Adygei a people of the Caucasus). Fourth the minority Mongoloid and "Boreal" (Purple) influence in Russians is affirmed. Note that we are dealing with ethnic Russians from the north (Vologda oblast), and Russians are a heterogeneous people in terms of their origin.
We can only wish for inclusion of further populations in future studies of the kind. In particular, eastern and southeastern Europe and non-Arab West Asia, Siberia would be invaluable in further understanding Caucasoid origins, and perhaps uncovering additional structure.
Note that the CEPH panel has revealed that ethnic groups can be distinguished genetically, there is little more than it can offer in terms of understanding origins. The next milestone would be either to include previously unsampled populations, or to dig into ethnic groups themselves, and see if sub-ethnic entities are also discernible in our genomes. Genetic genealogists are in for a good time in the coming years...
Science 22 February 2008: Vol. 319. no. 5866, pp. 1100 - 1104
Worldwide Human Relationships Inferred from Genome-Wide Patterns of Variation
Jun Z. Li,1,2*{dagger} Devin M. Absher,1,2* Hua Tang,1 Audrey M. Southwick,1,2 Amanda M. Casto,1 Sohini Ramachandran,4 Howard M. Cann,5 Gregory S. Barsh,1,3 Marcus Feldman,4{ddagger} Luigi L. Cavalli-Sforza,1{ddagger} Richard M. Myers1,2{ddagger}
Human genetic diversity is shaped by both demographic and biological factors and has fundamental implications for understanding the genetic basis of diseases. We studied 938 unrelated individuals from 51 populations of the Human Genome Diversity Panel at 650,000 common single-nucleotide polymorphism loci. Individual ancestry and population substructure were detectable with very high resolution. The relationship between haplotype heterozygosity and geography was consistent with the hypothesis of a serial founder effect with a single origin in sub-Saharan Africa. In addition, we observed a pattern of ancestral allele frequency distributions that reflects variation in population dynamics among geographic regions. This data set allows the most comprehensive characterization to date of human genetic variation.
Link
February 21, 2008
New study on global human variation based on SNPs and CNVs
A new letter in Nature combines data from single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) across 29 human populations. STRUCTURE results from the paper are below based on SNPs, haplotypes, and CNVs. Note in particular the Green cluster, which was not seen in some previous studies that did not include Oceanian populations, the differentiation between African farmers and hunter-gatherers, and the differentiation between northern and southern Mongoloids evident in the bottom row.
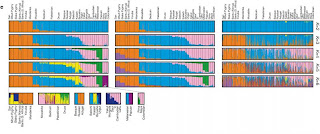
Nature 451, 998-1003 (21 February 2008) | doi:10.1038/nature06742; Received 2 December 2007; Accepted 29 January 2008
Genotype, haplotype and copy-number variation in worldwide human populations
Mattias Jakobsson1,2,14, Sonja W. Scholz4,5,14, Paul Scheet1,3,14, J. Raphael Gibbs4,5, Jenna M. VanLiere1, Hon-Chung Fung4,6, Zachary A. Szpiech1, James H. Degnan1,2, Kai Wang7, Rita Guerreiro4,8, Jose M. Bras4,8, Jennifer C. Schymick4,9, Dena G. Hernandez4, Bryan J. Traynor4,10, Javier Simon-Sanchez4,11, Mar Matarin4, Angela Britton4, Joyce van de Leemput4,5, Ian Rafferty4, Maja Bucan7, Howard M. Cann12, John A. Hardy5, Noah A. Rosenberg1,2,3 & Andrew B. Singleton4,13
Genome-wide patterns of variation across individuals provide a powerful source of data for uncovering the history of migration, range expansion, and adaptation of the human species. However, high-resolution surveys of variation in genotype, haplotype and copy number have generally focused on a small number of population groups1, 2, 3. Here we report the analysis of high-quality genotypes at 525,910 single-nucleotide polymorphisms (SNPs) and 396 copy-number-variable loci in a worldwide sample of 29 populations. Analysis of SNP genotypes yields strongly supported fine-scale inferences about population structure. Increasing linkage disequilibrium is observed with increasing geographic distance from Africa, as expected under a serial founder effect for the out-of-Africa spread of human populations. New approaches for haplotype analysis produce inferences about population structure that complement results based on unphased SNPs. Despite a difference from SNPs in the frequency spectrum of the copy-number variants (CNVs) detected—including a comparatively large number of CNVs in previously unexamined populations from Oceania and the Americas—the global distribution of CNVs largely accords with population structure analyses for SNP data sets of similar size. Our results produce new inferences about inter-population variation, support the utility of CNVs in human population-genetic research, and serve as a genomic resource for human-genetic studies in diverse worldwide populations.
Link

Nature 451, 998-1003 (21 February 2008) | doi:10.1038/nature06742; Received 2 December 2007; Accepted 29 January 2008
Genotype, haplotype and copy-number variation in worldwide human populations
Mattias Jakobsson1,2,14, Sonja W. Scholz4,5,14, Paul Scheet1,3,14, J. Raphael Gibbs4,5, Jenna M. VanLiere1, Hon-Chung Fung4,6, Zachary A. Szpiech1, James H. Degnan1,2, Kai Wang7, Rita Guerreiro4,8, Jose M. Bras4,8, Jennifer C. Schymick4,9, Dena G. Hernandez4, Bryan J. Traynor4,10, Javier Simon-Sanchez4,11, Mar Matarin4, Angela Britton4, Joyce van de Leemput4,5, Ian Rafferty4, Maja Bucan7, Howard M. Cann12, John A. Hardy5, Noah A. Rosenberg1,2,3 & Andrew B. Singleton4,13
Genome-wide patterns of variation across individuals provide a powerful source of data for uncovering the history of migration, range expansion, and adaptation of the human species. However, high-resolution surveys of variation in genotype, haplotype and copy number have generally focused on a small number of population groups1, 2, 3. Here we report the analysis of high-quality genotypes at 525,910 single-nucleotide polymorphisms (SNPs) and 396 copy-number-variable loci in a worldwide sample of 29 populations. Analysis of SNP genotypes yields strongly supported fine-scale inferences about population structure. Increasing linkage disequilibrium is observed with increasing geographic distance from Africa, as expected under a serial founder effect for the out-of-Africa spread of human populations. New approaches for haplotype analysis produce inferences about population structure that complement results based on unphased SNPs. Despite a difference from SNPs in the frequency spectrum of the copy-number variants (CNVs) detected—including a comparatively large number of CNVs in previously unexamined populations from Oceania and the Americas—the global distribution of CNVs largely accords with population structure analyses for SNP data sets of similar size. Our results produce new inferences about inter-population variation, support the utility of CNVs in human population-genetic research, and serve as a genomic resource for human-genetic studies in diverse worldwide populations.
Link
February 15, 2008
Pinghua Han Chinese are not of Han origin
An interesting study which demonstrates how a small subgroup of the Han Chinese, the Pinghua have a different origin than the rest of the Han. As in the case of some Finnish origin Russians, this illustrates that despite the general genetic coherence of ethnic groups, they can nonetheless include segments that were grafted onto their body. It is indeed the fact that ethnic groups constitute in a sense in many cases their own mini-races that allows us to identify outlier populations of different origins.
J Hum Genet. 2008 Feb 13 [Epub ahead of print]
Pinghua population as an exception of Han Chinese's coherent genetic structure.
Gan RJ et al.
The Han Chinese is the largest single ethnic group in the world, consisting of ten Chinese branches. With the exception of the Pinghua branch, the genetic structure of this group has been studied extensively, and Y chromosome and mitochondrial (mt)DNA data have demonstrated a coherent genetic structure of all Han Chinese. It is therefore believed that the Pinghua branch, being members of an old branch of the Han Chinese, despite being scattered in and around Guangxi Province where members of the Daic and Hmong-Mien are more prevalent than Han Chinese, is no exception. We have studied 470 individual samples (including 195 males) from Pinghua populations and other ethnic groups (Zhuang, Kam, Mulam, Laka, and Mien) from six areas (Hezhou, Fuchuan, Luocheng, Jinxiu, Sanjiang, and Wuxuan) in the north of the Guangxi Zhuang Autonomous Region of China. Both mtDNA and the Y chromosomes were typed in these samples. High frequencies of the Y chromosome haplogroups O2a* and O*, which always present at a high frequency among the populations of the southern minorities, were found in Pinghua populations. Only Pinghua populations in Luocheng and Jinxiu maintain the Han frequent haplogroup O3a5a. mtDNA lineages B4a, B5a, M*, F1a, M7b1, and N* were found in Pinghua populations, exhibiting a pattern similar to the neighboring indigenous populations, especially the Daic populations. Cluster analyses (dendrograms, principal component analyses, and networks) of Pinghua populations, the other Han branches, and other ethnic groups in East Asia indicated that Pinghua populations are much closer to the southern minorities than to the other Han branches. Admixture analyses confirmed this result. In conclusion, we argue that Pinghua populations did not descend from Han Chinese, but from southern minorities. The ancestral populations of Pinghua people were assimilated by the Han Chinese in terms of language, culture, and self-identification and, consequently, the Pinghua people became an exceptional branch of Han Chinese's coherent genetic structure.
Link
J Hum Genet. 2008 Feb 13 [Epub ahead of print]
Pinghua population as an exception of Han Chinese's coherent genetic structure.
Gan RJ et al.
The Han Chinese is the largest single ethnic group in the world, consisting of ten Chinese branches. With the exception of the Pinghua branch, the genetic structure of this group has been studied extensively, and Y chromosome and mitochondrial (mt)DNA data have demonstrated a coherent genetic structure of all Han Chinese. It is therefore believed that the Pinghua branch, being members of an old branch of the Han Chinese, despite being scattered in and around Guangxi Province where members of the Daic and Hmong-Mien are more prevalent than Han Chinese, is no exception. We have studied 470 individual samples (including 195 males) from Pinghua populations and other ethnic groups (Zhuang, Kam, Mulam, Laka, and Mien) from six areas (Hezhou, Fuchuan, Luocheng, Jinxiu, Sanjiang, and Wuxuan) in the north of the Guangxi Zhuang Autonomous Region of China. Both mtDNA and the Y chromosomes were typed in these samples. High frequencies of the Y chromosome haplogroups O2a* and O*, which always present at a high frequency among the populations of the southern minorities, were found in Pinghua populations. Only Pinghua populations in Luocheng and Jinxiu maintain the Han frequent haplogroup O3a5a. mtDNA lineages B4a, B5a, M*, F1a, M7b1, and N* were found in Pinghua populations, exhibiting a pattern similar to the neighboring indigenous populations, especially the Daic populations. Cluster analyses (dendrograms, principal component analyses, and networks) of Pinghua populations, the other Han branches, and other ethnic groups in East Asia indicated that Pinghua populations are much closer to the southern minorities than to the other Han branches. Admixture analyses confirmed this result. In conclusion, we argue that Pinghua populations did not descend from Han Chinese, but from southern minorities. The ancestral populations of Pinghua people were assimilated by the Han Chinese in terms of language, culture, and self-identification and, consequently, the Pinghua people became an exceptional branch of Han Chinese's coherent genetic structure.
Link
News on Arabian mtDNA
Not one but two recent paper on Arabian mtDNA, giving us a better idea of its geographical structure. I am not sure what to make of the assertion in the first paper that the Arabian peninsula has been the recipient of genetic input from Australia; well, it's in an open access journal so you can form your own opinions.
BMC Evol Biol. 2008 Feb 12;8(1):45 [Epub ahead of print]
Mitochondrial DNA structure in the Arabian Peninsula.
Abu-Amero KK, Larruga JM, Cabrera VM, Gonzalez AM.
ABSTRACT: BACKGROUND: Two potential migratory routes followed by modern humans to colonize Eurasia from Africa have been proposed. These are the two natural passageways that connect both continents: the northern route through the Sinai Peninsula and the southern route across the Bab al Mandab strait. Recent archaeological and genetic evidence have favored a unique southern coastal route. Under this scenario, the study of the population genetic structure of the Arabian Peninsula, the first step out of Africa, to search for primary genetic links between Africa and Eurasia, is crucial. The haploid and maternally inherited mitochondrial DNA (mtDNA) molecule has been the most used genetic marker to identify and to relate lineages with clear geographic origins, as the African Ls and the Eurasian M and N that have a common root with the Africans L3. RESULTS: To assess the role of the Arabian Peninsula in the southern route, we genetically analyzed 553 Saudi Arabs using partial (546) and complete mtDNA (7) sequencing, and compared the lineages obtained with those present in Africa, the Near East, central, east and southeast Asia and Australasia. The results showed that the Arabian Peninsula has received substantial gene flow from Africa (20%), detected by the presence of L, M1 and U6 lineages; that an 18% of the Arabian Peninsula lineages have a clear eastern provenance, mainly represented by U lineages; but also by Indian M lineages and rare M links with Central Asia, Indonesia and even Australia. However, the bulk (62%) of the Arabian lineages has a Northern source. CONCLUSIONS: Although there is evidence of Neolithic and more recent expansions in the Arabian Peninsula, mainly detected by (preHV)1 and J1b lineages, the lack of primitive autochthonous M and N sequences, suggests that this area has been more a receptor of human migrations, including historic ones, from Africa, India, Indonesia and even Australia, than a demographic expansion center along the proposed southern coastal route.
Link
Am J Phys Anthropol. 2008 Feb 6 [Epub ahead of print]
Regional differences in the distribution of the sub-Saharan, West Eurasian, and South Asian mtDNA lineages in Yemen.
Cerný V et al.
Despite its key location for population movements out of and back into Africa, Yemen has not yet been sampled on a regional level for an investigation of sub-Saharan, West Eurasian, and South Asian genetic contributions. In this study, we present mitochondrial DNA (mtDNA) data for regionally distinct Yemeni populations that reveal different distributions of mtDNA lineages. An extensive database of mtDNA sequences from North and East African, Middle Eastern and Indian populations was analyzed to provide a context for the regional Yemeni mtDNA datasets. The groups of western Yemen appear to be most closely related to Middle Eastern and North African populations, while the eastern Yemeni population from Hadramawt is most closely related to East Africa. Furthermore, haplotype matches with Africa are almost exclusively confined to West Eurasian R0a haplogroup in southwestern Yemen, although more sub-Saharan L-type matches appear in more northern Yemeni populations. In fact, Yemeni populations have the highest frequency of R0a haplotypes detected to date, thus Yemen or southern Arabia may be the site of the initial expansion of this haplogroup. Whereas two variants of the sub-Saharan haplogroup M1 were detected only in southwestern Yemen close to the Bab el-Mandeb Strait, different non-African M haplotypes were detected at low frequencies ( approximately 2%) in western parts of the country and at a higher frequency (7.5%) in the Hadramawt. We conclude that the Yemeni gene pool is highly stratified both regionally and temporally and that it has received West Eurasian, Northeast African, and South Asian gene flow.
Link
BMC Evol Biol. 2008 Feb 12;8(1):45 [Epub ahead of print]
Mitochondrial DNA structure in the Arabian Peninsula.
Abu-Amero KK, Larruga JM, Cabrera VM, Gonzalez AM.
ABSTRACT: BACKGROUND: Two potential migratory routes followed by modern humans to colonize Eurasia from Africa have been proposed. These are the two natural passageways that connect both continents: the northern route through the Sinai Peninsula and the southern route across the Bab al Mandab strait. Recent archaeological and genetic evidence have favored a unique southern coastal route. Under this scenario, the study of the population genetic structure of the Arabian Peninsula, the first step out of Africa, to search for primary genetic links between Africa and Eurasia, is crucial. The haploid and maternally inherited mitochondrial DNA (mtDNA) molecule has been the most used genetic marker to identify and to relate lineages with clear geographic origins, as the African Ls and the Eurasian M and N that have a common root with the Africans L3. RESULTS: To assess the role of the Arabian Peninsula in the southern route, we genetically analyzed 553 Saudi Arabs using partial (546) and complete mtDNA (7) sequencing, and compared the lineages obtained with those present in Africa, the Near East, central, east and southeast Asia and Australasia. The results showed that the Arabian Peninsula has received substantial gene flow from Africa (20%), detected by the presence of L, M1 and U6 lineages; that an 18% of the Arabian Peninsula lineages have a clear eastern provenance, mainly represented by U lineages; but also by Indian M lineages and rare M links with Central Asia, Indonesia and even Australia. However, the bulk (62%) of the Arabian lineages has a Northern source. CONCLUSIONS: Although there is evidence of Neolithic and more recent expansions in the Arabian Peninsula, mainly detected by (preHV)1 and J1b lineages, the lack of primitive autochthonous M and N sequences, suggests that this area has been more a receptor of human migrations, including historic ones, from Africa, India, Indonesia and even Australia, than a demographic expansion center along the proposed southern coastal route.
Link
Am J Phys Anthropol. 2008 Feb 6 [Epub ahead of print]
Regional differences in the distribution of the sub-Saharan, West Eurasian, and South Asian mtDNA lineages in Yemen.
Cerný V et al.
Despite its key location for population movements out of and back into Africa, Yemen has not yet been sampled on a regional level for an investigation of sub-Saharan, West Eurasian, and South Asian genetic contributions. In this study, we present mitochondrial DNA (mtDNA) data for regionally distinct Yemeni populations that reveal different distributions of mtDNA lineages. An extensive database of mtDNA sequences from North and East African, Middle Eastern and Indian populations was analyzed to provide a context for the regional Yemeni mtDNA datasets. The groups of western Yemen appear to be most closely related to Middle Eastern and North African populations, while the eastern Yemeni population from Hadramawt is most closely related to East Africa. Furthermore, haplotype matches with Africa are almost exclusively confined to West Eurasian R0a haplogroup in southwestern Yemen, although more sub-Saharan L-type matches appear in more northern Yemeni populations. In fact, Yemeni populations have the highest frequency of R0a haplotypes detected to date, thus Yemen or southern Arabia may be the site of the initial expansion of this haplogroup. Whereas two variants of the sub-Saharan haplogroup M1 were detected only in southwestern Yemen close to the Bab el-Mandeb Strait, different non-African M haplotypes were detected at low frequencies ( approximately 2%) in western parts of the country and at a higher frequency (7.5%) in the Hadramawt. We conclude that the Yemeni gene pool is highly stratified both regionally and temporally and that it has received West Eurasian, Northeast African, and South Asian gene flow.
Link
February 14, 2008
New synthesis on the first arrivals into the New World
In the open access journal PLoS One.
A Three-Stage Colonization Model for the Peopling of the Americas
Andrew Kitchen et al.
Abstract
Background
We evaluate the process by which the Americas were originally colonized and propose a three-stage model that integrates current genetic, archaeological, geological, and paleoecological data. Specifically, we analyze mitochondrial and nuclear genetic data by using complementary coalescent models of demographic history and incorporating non-genetic data to enhance the anthropological relevance of the analysis.
Methodology/Findings
Bayesian skyline plots, which provide dynamic representations of population size changes over time, indicate that Amerinds went through two stages of growth ≈40,000 and ≈15,000 years ago separated by a long period of population stability. Isolation-with-migration coalescent analyses, which utilize data from sister populations to estimate a divergence date and founder population sizes, suggest an Amerind population expansion starting ≈15,000 years ago.
Conclusions/Significance
These results support a model for the peopling of the New World in which Amerind ancestors diverged from the Asian gene pool prior to 40,000 years ago and experienced a gradual population expansion as they moved into Beringia. After a long period of little change in population size in greater Beringia, Amerinds rapidly expanded into the Americas ≈15,000 years ago either through an interior ice-free corridor or along the coast. This rapid colonization of the New World was achieved by a founder group with an effective population size of ≈1,000–5,400 individuals. Our model presents a detailed scenario for the timing and scale of the initial migration to the Americas, substantially refines the estimate of New World founders, and provides a unified theory for testing with future datasets and analytic methods.
Link
A Three-Stage Colonization Model for the Peopling of the Americas
Andrew Kitchen et al.
Abstract
Background
We evaluate the process by which the Americas were originally colonized and propose a three-stage model that integrates current genetic, archaeological, geological, and paleoecological data. Specifically, we analyze mitochondrial and nuclear genetic data by using complementary coalescent models of demographic history and incorporating non-genetic data to enhance the anthropological relevance of the analysis.
Methodology/Findings
Bayesian skyline plots, which provide dynamic representations of population size changes over time, indicate that Amerinds went through two stages of growth ≈40,000 and ≈15,000 years ago separated by a long period of population stability. Isolation-with-migration coalescent analyses, which utilize data from sister populations to estimate a divergence date and founder population sizes, suggest an Amerind population expansion starting ≈15,000 years ago.
Conclusions/Significance
These results support a model for the peopling of the New World in which Amerind ancestors diverged from the Asian gene pool prior to 40,000 years ago and experienced a gradual population expansion as they moved into Beringia. After a long period of little change in population size in greater Beringia, Amerinds rapidly expanded into the Americas ≈15,000 years ago either through an interior ice-free corridor or along the coast. This rapid colonization of the New World was achieved by a founder group with an effective population size of ≈1,000–5,400 individuals. Our model presents a detailed scenario for the timing and scale of the initial migration to the Americas, substantially refines the estimate of New World founders, and provides a unified theory for testing with future datasets and analytic methods.
Link
February 11, 2008
Maternal common ancestry between pygmies and farmers in Central Africa
This is an Open Access article in PNAS.
PNAS 10.1073/pnas.0711467105
Maternal traces of deep common ancestry and asymmetric gene flow between Pygmy hunter–gatherers and Bantu-speaking farmers
Lluís Quintana-Murci et al.
Abstract
Two groups of populations with completely different lifestyles—the Pygmy hunter–gatherers and the Bantu-speaking farmers—coexist in Central Africa. We investigated the origins of these two groups and the interactions between them, by analyzing mtDNA variation in 1,404 individuals from 20 farming populations and 9 Pygmy populations from Central Africa, with the aim of shedding light on one of the most fascinating cultural transitions in human evolution (the transition from hunting and gathering to agriculture). Our data indicate that this region was colonized gradually, with an initial L1c-rich ancestral population ultimately giving rise to current-day farmers, who display various L1c clades, and to Pygmies, in whom L1c1a is the only surviving clade. Detailed phylogenetic analysis of complete mtDNA sequences for L1c1a showed this clade to be autochthonous to Central Africa, with its most recent branches shared between farmers and Pygmies. Coalescence analyses revealed that these two groups arose through a complex evolutionary process characterized by (i) initial divergence of the ancestors of contemporary Pygmies from an ancestral Central African population no more than {approx}70,000 years ago, (ii) a period of isolation between the two groups, accounting for their phenotypic differences, (iii) long-standing asymmetric maternal gene flow from Pygmies to the ancestors of the farming populations, beginning no more than {approx}40,000 years ago and persisting until a few thousand years ago, and (iv) enrichment of the maternal gene pool of the ancestors of the farming populations by the arrival and/or subsequent demographic expansion of L0a, L2, and L3 carriers.
Link
PNAS 10.1073/pnas.0711467105
Maternal traces of deep common ancestry and asymmetric gene flow between Pygmy hunter–gatherers and Bantu-speaking farmers
Lluís Quintana-Murci et al.
Abstract
Two groups of populations with completely different lifestyles—the Pygmy hunter–gatherers and the Bantu-speaking farmers—coexist in Central Africa. We investigated the origins of these two groups and the interactions between them, by analyzing mtDNA variation in 1,404 individuals from 20 farming populations and 9 Pygmy populations from Central Africa, with the aim of shedding light on one of the most fascinating cultural transitions in human evolution (the transition from hunting and gathering to agriculture). Our data indicate that this region was colonized gradually, with an initial L1c-rich ancestral population ultimately giving rise to current-day farmers, who display various L1c clades, and to Pygmies, in whom L1c1a is the only surviving clade. Detailed phylogenetic analysis of complete mtDNA sequences for L1c1a showed this clade to be autochthonous to Central Africa, with its most recent branches shared between farmers and Pygmies. Coalescence analyses revealed that these two groups arose through a complex evolutionary process characterized by (i) initial divergence of the ancestors of contemporary Pygmies from an ancestral Central African population no more than {approx}70,000 years ago, (ii) a period of isolation between the two groups, accounting for their phenotypic differences, (iii) long-standing asymmetric maternal gene flow from Pygmies to the ancestors of the farming populations, beginning no more than {approx}40,000 years ago and persisting until a few thousand years ago, and (iv) enrichment of the maternal gene pool of the ancestors of the farming populations by the arrival and/or subsequent demographic expansion of L0a, L2, and L3 carriers.
Link
Third cousin marriage and maximum fertility
John Hawks and Steve Sailer discuss a new Science paper (Agnar Helgason et al., "An Association Between the Kinship and Fertility of Human Couples", Science 8 February 2008: Vol. 319. no. 5864, pp. 813 - 816) on the correlation between fertility and degree of kinship in Iceland. From the public release about the paper:
One explanation is that there is some biological factor which decreases fitness as genetic dissimilarity increases. Perhaps more distantly related genomes don't "mesh" that well.
I suggest that the phenomenon could be partially explained by the fact that as kinship decreases, the age distribution of potential marriage partners becomes more varied.
A man's age difference from his sister is constrained by the fact that one's mother has a limited reproductive age range. In other words, one's sister is usually a few years older or younger than oneself, and rarely much older or younger.
But, if we look at first or second cousins, this difference increases. Your uncle or aunt is younger or older than your parents, and correspondingly their children can be more different than you in age.
As kinship decreases, your n-degree cousins become less constrained to be close to you in age. They could be much much older (if they are descended from short branches of one's family tree), or much much younger (if they are descended from long ones).
Of course, men usually marry women who are not that different from them in age (see data for Norway). But, the opportunity to marry someone much younger (or much older) than oneself is greater as kinship decreases.
It would be interesting to see data on average age differences correlated with degree of kinship in Iceland. If it turns out that, say, third-cousin marriage partners are less different in age than fourth-cousin partners, then an alternative explanation may be behind the observed fertility curve: age difference among spouses is negatively correlated with fertility.
In a paper published today deCODE scientists establish a substantial and consistent positive correlation between the kinship of couples and the number of children and grandchildren they have. The study, which analyzes more than 200 years of deCODE’s comprehensive genalogical data on the population of Iceland, shows that couples related at the level of third cousins have the greatest number of offspring.It is of course clear why close relatives (closer than 3rd cousins) should have fewer children: this is due to the well known phenomenon of inbreeding depression. However, why should fertility increase up to a certain degree of kinship (~3rd cousins) and decrease after that?
One explanation is that there is some biological factor which decreases fitness as genetic dissimilarity increases. Perhaps more distantly related genomes don't "mesh" that well.
I suggest that the phenomenon could be partially explained by the fact that as kinship decreases, the age distribution of potential marriage partners becomes more varied.
A man's age difference from his sister is constrained by the fact that one's mother has a limited reproductive age range. In other words, one's sister is usually a few years older or younger than oneself, and rarely much older or younger.
But, if we look at first or second cousins, this difference increases. Your uncle or aunt is younger or older than your parents, and correspondingly their children can be more different than you in age.
As kinship decreases, your n-degree cousins become less constrained to be close to you in age. They could be much much older (if they are descended from short branches of one's family tree), or much much younger (if they are descended from long ones).
Of course, men usually marry women who are not that different from them in age (see data for Norway). But, the opportunity to marry someone much younger (or much older) than oneself is greater as kinship decreases.
It would be interesting to see data on average age differences correlated with degree of kinship in Iceland. If it turns out that, say, third-cousin marriage partners are less different in age than fourth-cousin partners, then an alternative explanation may be behind the observed fertility curve: age difference among spouses is negatively correlated with fertility.
February 10, 2008
New paper on genomic differences between Ashkenazi Jews and Europeans
A reader pointed me to a new paper in the open access BMC Genetics journal. I won't comment on the main subject matter of the paper, but the PC scatterplot confirms some of the work I posted about previously (and here) with regard to the distinctiveness of Ashkenazi Jewish gene pool. With the exception of a single AJ individual falling in the CEU cluster (on the right), there is a clean separation between the two clusters. Note also how surprisingly tight the two clusters are; perhaps the AJ outliers (marked with arrows) may be due to some non-AJ ancestry. With 500,000 SNPs we are beginning to move into ethnic genetics territory, where not only race and subrace but also ethnicity may become genetically traceable.
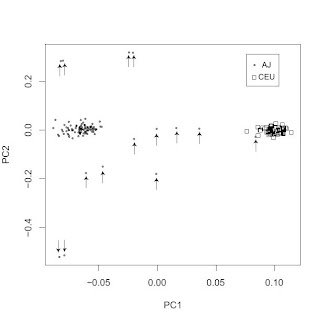
ADDENDUM:
A common misconception, which I hear about often when people are exposed to PC plots such as the above is that it is taken as evidence for a lack or shortage of gene exchange between the clearly separated populations.
This is not, however, the case. What the plot shows is that you can score a person's genome on two variables (the first two principal components) giving you an (x, y) pair. Using this pair, you can guess confidently that they are much more likely to be of AJ rather than CEU origin or vice versa.
This does not mean that there has been limited gene flow between AJs and CEUs. It just means that historically intermarriage did not win out against intramarriage, and hence AJs and CEUs could develop distinctive gene pools.
BMC Genetics
Analysis of genetic variation in Ashkenazi Jews by high density SNP genotyping
Adam B Olshen et al.
Abstract (provisional)
Background
Genetic isolates such as the Ashkenazi Jews (AJ) potentially offer advantages in mapping novel loci in whole genome disease association studies. To analyze patterns of genetic variation in AJ, genotypes of 101 healthy individuals were determined using the Affymetrix EAv3 500K SNP array and compared to 60 CEPH-derived HapMap (CEU) individuals. 435,632 SNPs overlapped and met annotation criteria in the two groups.
Results
A small but significant global difference in allele frequencies between AJ and CEU was demonstrated by a mean FST of 0.009 (P < style="font-weight: bold;">
Conclusions
LD in the AJ versus CEU was lower than expected by some measures and higher by others. Any putative advantage in whole genome association mapping using the AJ population will be highly dependent on regional LD structure.
Link (provisional pdf)

ADDENDUM:
A common misconception, which I hear about often when people are exposed to PC plots such as the above is that it is taken as evidence for a lack or shortage of gene exchange between the clearly separated populations.
This is not, however, the case. What the plot shows is that you can score a person's genome on two variables (the first two principal components) giving you an (x, y) pair. Using this pair, you can guess confidently that they are much more likely to be of AJ rather than CEU origin or vice versa.
This does not mean that there has been limited gene flow between AJs and CEUs. It just means that historically intermarriage did not win out against intramarriage, and hence AJs and CEUs could develop distinctive gene pools.
BMC Genetics
Analysis of genetic variation in Ashkenazi Jews by high density SNP genotyping
Adam B Olshen et al.
Abstract (provisional)
Background
Genetic isolates such as the Ashkenazi Jews (AJ) potentially offer advantages in mapping novel loci in whole genome disease association studies. To analyze patterns of genetic variation in AJ, genotypes of 101 healthy individuals were determined using the Affymetrix EAv3 500K SNP array and compared to 60 CEPH-derived HapMap (CEU) individuals. 435,632 SNPs overlapped and met annotation criteria in the two groups.
Results
A small but significant global difference in allele frequencies between AJ and CEU was demonstrated by a mean FST of 0.009 (P < style="font-weight: bold;">
Conclusions
LD in the AJ versus CEU was lower than expected by some measures and higher by others. Any putative advantage in whole genome association mapping using the AJ population will be highly dependent on regional LD structure.
Link (provisional pdf)
February 05, 2008
Y chromosomes and Origins of Cretan and mainland Greek Neolithic
This is an important new paper about Y chromosomes in Greece. The authors studied Cretans as well as Greek populations from early Neolithic sites in Macedonia, Thessaly, and the Peloponnese.
My main gripe -which is a general one and not limited to this paper- is for researchers to consider prehistorical explanations, related to "hot topics" in archaeology, such as the origin of the Neolithic, rather than more mundane historical ones.
It would be interesting to include a discussion of the post-Neolithic population settlement history of the studied areas: the idea that the current gene pools preserve a strong signal of the Neolithic inhabitants of the same areas seems rather arbitrary to me.
I will add more comments as I read the paper and supplementary information more closely, but one of the certain conclusions from the paper is that it places the final tombstone to the whole Bernal/Black Athena (or alternatively Poe/Black Spark, White Fire) theory of Aegean prehistory. The complete dissimilarity of E3b related lineages between Greece and Crete on the one hand and Egypt on the other, makes any type of Bronze Age colonization of the Aegean by Egyptians an impossibility.
Annals of Human Genetics, Volume 72 Issue 2 Page 205-214, March 2008
Differential Y-chromosome Anatolian Influences on the Greek and Cretan Neolithic
R. J. King et al.
The earliest Neolithic sites of Europe are located in Crete and mainland Greece. A debate persists concerning whether these farmers originated in neighboring Anatolia and the role of maritime colonization. To address these issues 171 samples were collected from areas near three known early Neolithic settlements in Greece together with 193 samples from Crete. An analysis of Y-chromosome haplogroups determined that the samples from the Greek Neolithic sites showed strong affinity to Balkan data, while Crete shows affinity with central/Mediterranean Anatolia. Haplogroup J2b-M12 was frequent in Thessaly and Greek Macedonia while haplogroup J2a-M410 was scarce. Alternatively, Crete, like Anatolia showed a high frequency of J2a-M410 and a low frequency of J2b-M12. This dichotomy parallels archaeobotanical evidence, specifically that while bread wheat (Triticum aestivum) is known from Neolithic Anatolia, Crete and southern Italy; it is absent from earliest Neolithic Greece. The expansion time of YSTR variation for haplogroup E3b1a2-V13, in the Peloponnese was consistent with an indigenous Mesolithic presence. In turn, two distinctive haplogroups, J2a1h-M319 and J2a1b1-M92, have demographic properties consistent with Bronze Age expansions in Crete, arguably from NW/W Anatolia and Syro-Palestine, while a later mainland (Mycenaean) contribution to Crete is indicated by relative frequencies of V13.
Link
My main gripe -which is a general one and not limited to this paper- is for researchers to consider prehistorical explanations, related to "hot topics" in archaeology, such as the origin of the Neolithic, rather than more mundane historical ones.
It would be interesting to include a discussion of the post-Neolithic population settlement history of the studied areas: the idea that the current gene pools preserve a strong signal of the Neolithic inhabitants of the same areas seems rather arbitrary to me.
I will add more comments as I read the paper and supplementary information more closely, but one of the certain conclusions from the paper is that it places the final tombstone to the whole Bernal/Black Athena (or alternatively Poe/Black Spark, White Fire) theory of Aegean prehistory. The complete dissimilarity of E3b related lineages between Greece and Crete on the one hand and Egypt on the other, makes any type of Bronze Age colonization of the Aegean by Egyptians an impossibility.
Annals of Human Genetics, Volume 72 Issue 2 Page 205-214, March 2008
Differential Y-chromosome Anatolian Influences on the Greek and Cretan Neolithic
R. J. King et al.
The earliest Neolithic sites of Europe are located in Crete and mainland Greece. A debate persists concerning whether these farmers originated in neighboring Anatolia and the role of maritime colonization. To address these issues 171 samples were collected from areas near three known early Neolithic settlements in Greece together with 193 samples from Crete. An analysis of Y-chromosome haplogroups determined that the samples from the Greek Neolithic sites showed strong affinity to Balkan data, while Crete shows affinity with central/Mediterranean Anatolia. Haplogroup J2b-M12 was frequent in Thessaly and Greek Macedonia while haplogroup J2a-M410 was scarce. Alternatively, Crete, like Anatolia showed a high frequency of J2a-M410 and a low frequency of J2b-M12. This dichotomy parallels archaeobotanical evidence, specifically that while bread wheat (Triticum aestivum) is known from Neolithic Anatolia, Crete and southern Italy; it is absent from earliest Neolithic Greece. The expansion time of YSTR variation for haplogroup E3b1a2-V13, in the Peloponnese was consistent with an indigenous Mesolithic presence. In turn, two distinctive haplogroups, J2a1h-M319 and J2a1b1-M92, have demographic properties consistent with Bronze Age expansions in Crete, arguably from NW/W Anatolia and Syro-Palestine, while a later mainland (Mycenaean) contribution to Crete is indicated by relative frequencies of V13.
Link
Temperature and sex ratio
This is an open-access article, so you can read it in its entirety.
Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0710711104
Ambient temperature predicts sex ratios and male longevity
Ralph Catalano et al.
Abstract
The theory that natural selection has conserved mechanisms by which women subjected to environmental stressors abort frail male fetuses implies that climate change may affect sex ratio at birth and male longevity. Using time series methods, we find that cold ambient temperatures during gestation predict lower secondary sex ratios and longer life span of males in annual birth cohorts composed of Danes, Finns, Norwegians, and Swedes born between 1878 (earliest year with complete life tables) and 1914 (last birth cohort for which male life span can be estimated). We conclude that ambient temperature affects the characteristics of human populations by influencing who survives gestation, a heretofore unrecognized effect of climate on humanity.
Link
Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0710711104
Ambient temperature predicts sex ratios and male longevity
Ralph Catalano et al.
Abstract
The theory that natural selection has conserved mechanisms by which women subjected to environmental stressors abort frail male fetuses implies that climate change may affect sex ratio at birth and male longevity. Using time series methods, we find that cold ambient temperatures during gestation predict lower secondary sex ratios and longer life span of males in annual birth cohorts composed of Danes, Finns, Norwegians, and Swedes born between 1878 (earliest year with complete life tables) and 1914 (last birth cohort for which male life span can be estimated). We conclude that ambient temperature affects the characteristics of human populations by influencing who survives gestation, a heretofore unrecognized effect of climate on humanity.
Link
February 04, 2008
South American mtDNA, re-interpretation of its prehistory
I really love this type of paper that focuses on methodological criticism of population genetics work.
Molecular Biology and Evolution, doi:10.1093/molbev/msm225
Native South American Genetic Structure and Prehistory Inferred from Hierarchical Modeling of mtDNA
Cecil M. Lewis, Jr et al.
Genetic diversity in Native South Americans forms a complex pattern at both the continental and local levels. In comparing the West to the East, there is more variation within groups and smaller genetic distances between groups. From this pattern, researchers have proposed that that there is more variation in the West, and that a larger, more genetically diverse, founding population entered the West than the East. Here, we question this characterization of South American genetic variation, and its interpretation. Our concern arises because others have inferred regional variation from the mean variation within local populations without taking into account the variation among local populations within the same region. This failure produces a biased view of the actual variation in the East.
Link
Molecular Biology and Evolution, doi:10.1093/molbev/msm225
Native South American Genetic Structure and Prehistory Inferred from Hierarchical Modeling of mtDNA
Cecil M. Lewis, Jr et al.
Genetic diversity in Native South Americans forms a complex pattern at both the continental and local levels. In comparing the West to the East, there is more variation within groups and smaller genetic distances between groups. From this pattern, researchers have proposed that that there is more variation in the West, and that a larger, more genetically diverse, founding population entered the West than the East. Here, we question this characterization of South American genetic variation, and its interpretation. Our concern arises because others have inferred regional variation from the mean variation within local populations without taking into account the variation among local populations within the same region. This failure produces a biased view of the actual variation in the East.
Link
February 01, 2008
300K SNP paper on European genetic substructure

This is another recent paper using a large number of genetic markers to study genetic structure within a race. You can read the paper for yourselves, but some comments are in order.
First, this mirrors the results of another recent study on European-American population structure. Mediterranean populations are distinguished from Northern European populations and from Jews.
Doubtlessly, the discontinuities between the three groups evident in the Figure would be less clear if more populations were sampled, but nonetheless this is clear evidence that not only race, but ethnicity too has a genetic component.
If e.g., we were given the DNA of a German, a Greek, and a Jew, we would be able to tell them apart with very high probability. All it would take, would be to genotype them for the 300K SNPs, and subsequently score them for the first two principal components.
Another interesting feature of this study is the form of the Jewish cluster. The purple dots correspond to Jewsh with all four Jewish grandparents and they form a tight cluster. This is not the case when less stringent criteria (blue dots) are used.
This is a wonderful visual display of what happens to a partially inbred group (Ashkenazi Jews) as a result of intermixture. As long as within-group marriage patterns prevail, genetic similarity is maintained, but intermarriage leads to an attenuation of the group's genetic distinctiveness.
It is interesting to think of how this picture will change as genetic science moves forward:
If more populations are sampled, then the gaps between populations may "fill up". I would not be surprised, for example, if the French would occupy the gap between Spaniards and Germans.
If more SNPs were used, then populations that overlap greatly (say Greeks and Italians) might be separated.
Ethnic groups may differ both in culture and in genes, but these two dimensions of ethnicity are not independent. Differences in culture are barriers to gene flow; over time, such barriers differentiate gene pools. Conversely, genetic intermixture bring separate gene pools closer together, but also serves to homogenize cultures.
PLoS Genetics Vol. 4, No. 1, e4 doi:10.1371/journal.pgen.0040004
Analysis and Application of European Genetic Substructure Using 300 K SNP Information
Tian C, Plenge RM, Ransom M, Lee A, Villoslada P, et al.
European population genetic substructure was examined in a diverse set of >1,000 individuals of European descent, each genotyped with >300 K SNPs. Both STRUCTURE and principal component analyses (PCA) showed the largest division/principal component (PC) differentiated northern from southern European ancestry. A second PC further separated Italian, Spanish, and Greek individuals from those of Ashkenazi Jewish ancestry as well as distinguishing among northern European populations. In separate analyses of northern European participants other substructure relationships were discerned showing a west to east gradient. Application of this substructure information was critical in examining a real dataset in whole genome association (WGA) analyses for rheumatoid arthritis in European Americans to reduce false positive signals. In addition, two sets of European substructure ancestry informative markers (ESAIMs) were identified that provide substantial substructure information. The results provide further insight into European population genetic substructure and show that this information can be used for improving error rates in association testing of candidate genes and in replication studies of WGA scans.
Link
Subscribe to:
Posts (Atom)