Of wider interest might be the authors' estimation of the autosomal mutation rate as 1.44x10-8 mutations/bp/generation. Of course, this might depend on the archaeological calibration used (where/when did the bottleneck in the ancestry of Native Americans occur?). It might also depend on recent evidence that Native Americans are of mixed origin and thus did not really split from CHB/JPT; only part of their ancestry did. Nonetheless, this is another fairly "low" autosomal mutation rate.
(This was previously released as a preprint to the arXiv).
PLoS Genet 9(12): e1004023. doi:10.1371/journal.pgen.1004023
Reconstructing Native American Migrations from Whole-Genome and Whole-Exome Data
Simon Gravel et al.
Link

December 27, 2013
Site frequency spectrum from reads is unbiased (from genotype calls, biased at low coverage)
Mol Biol Evol (2013)
doi: 10.1093/molbev/mst229
Characterizing Bias in Population Genetic Inferences from Low-Coverage Sequencing Data
Eunjung Han et al.
The site frequency spectrum (SFS) is of primary interest in population genetic studies, because the SFS compresses variation data into a simple summary from which many population genetic inferences can proceed. However, inferring the SFS from sequencing data is challenging because genotype calls from sequencing data are often inaccurate due to high error rates and if not accounted for, this genotype uncertainty can lead to serious bias in downstream analysis based on the inferred SFS. Here, we compare two approaches to estimate the SFS from sequencing data: one approach infers individual genotypes from aligned sequencing reads and then estimates the SFS based on the inferred genotypes (call-based approach) and the other approach directly estimates the SFS from aligned sequencing reads by maximum likelihood (direct estimation approach). We find that the SFS estimated by the direct estimation approach is unbiased even at low coverage, whereas the SFS by the call-based approach becomes biased as coverage decreases. The direction of the bias in the call-based approach depends on the pipeline to infer genotypes. Estimating genotypes by pooling individuals in a sample (multisample calling) results in underestimation of the number of rare variants, whereas estimating genotypes in each individual and merging them later (single-sample calling) leads to overestimation of rare variants. We characterize the impact of these biases on downstream analyses, such as demographic parameter estimation and genome-wide selection scans. Our work highlights that depending on the pipeline used to infer the SFS, one can reach different conclusions in population genetic inference with the same data set. Thus, careful attention to the analysis pipeline and SFS estimation procedures is vital for population genetic inferences.
Link
Characterizing Bias in Population Genetic Inferences from Low-Coverage Sequencing Data
Eunjung Han et al.
The site frequency spectrum (SFS) is of primary interest in population genetic studies, because the SFS compresses variation data into a simple summary from which many population genetic inferences can proceed. However, inferring the SFS from sequencing data is challenging because genotype calls from sequencing data are often inaccurate due to high error rates and if not accounted for, this genotype uncertainty can lead to serious bias in downstream analysis based on the inferred SFS. Here, we compare two approaches to estimate the SFS from sequencing data: one approach infers individual genotypes from aligned sequencing reads and then estimates the SFS based on the inferred genotypes (call-based approach) and the other approach directly estimates the SFS from aligned sequencing reads by maximum likelihood (direct estimation approach). We find that the SFS estimated by the direct estimation approach is unbiased even at low coverage, whereas the SFS by the call-based approach becomes biased as coverage decreases. The direction of the bias in the call-based approach depends on the pipeline to infer genotypes. Estimating genotypes by pooling individuals in a sample (multisample calling) results in underestimation of the number of rare variants, whereas estimating genotypes in each individual and merging them later (single-sample calling) leads to overestimation of rare variants. We characterize the impact of these biases on downstream analyses, such as demographic parameter estimation and genome-wide selection scans. Our work highlights that depending on the pipeline used to infer the SFS, one can reach different conclusions in population genetic inference with the same data set. Thus, careful attention to the analysis pipeline and SFS estimation procedures is vital for population genetic inferences.
Link
December 26, 2013
Ancient DNA: what 2013 has brought
I was looking at my ancient DNA tag for the last year and it seems we've learned quite a lot in 2013. Here's my short summary of some major studies, news articles and reports:
- 400,000 year old Homo heidelbergensis in Iberia had mtDNA similar to Middle Paleolithic Denisovans from the Altai. This is important because of the age of the sample which opens up new vistas for ancient DNA research and because it is the first link to the mysterious Denisovans.
- A Neandertal inhabited the same cave where the Denisovan fingerbone was found. Denisovans had Neandertal admixture as well as admixture with an unknown "ultra-archaic" group; Eurasians have admixture from a Neandertal most similar to the Mezmaiskaya sample from the Caucasus; East Eurasians have a little bit of Denisovan admixture, while Australasians have a lot more; and all Sub-Saharan Africans seem to have a little bit of Neandertal admixture too, via West Eurasians during the Holocene.
- A ~24,000 year old Upper Paleolithic Siberian from Mal'ta is related to Native Americans who are a mix of it and East Asians. Mal'ta was related to West Eurasians and not to East Eurasians. It belonged to Y-haplogroup R* and mt-haplogroup U*.
- On the other hand, a ~40,000 year old from China was definitely East Eurasian.
- Europeans are a 3-way mix of Neolithic farmers, Mesolithic hunter-gatherers and aforementioned UP Siberian-like "Ancient North Eurasians"; Early LBK farmers from Central Europe resemble later Oetzi, Swedish farmers, and probably Iberian farmers too. They also had mysterious "Basal Eurasian" ancestry from the deepest split of the Eurasian tree. Mesolithic Europeans had lots of Y-haplogroup I.
- Ancient mtDNA reveals that something happened during the late Neolithic and early Bronze Age in Germany; the populations from that time are the first ones who appear quasi-modern in their haplogroup frequencies. It also turns out that hunter-gatherers didn't disappear in Germany after the LBK came along. And mtDNA haplogroup H, most frequent in modern Europeans, established itself around the Mid-to-Late Neolithic.
- West Siberia had a West/East Eurasian admixed population during the Bronze Age, like earlier ages.
- Lots of hints of interesting events in the European steppe too.
- Modern Tuscans probably not descended from ancient Etruscans; discontinuity seems to be the rule.
- Minoans were fairly regular Europeans, not North African (they had little mtDNA U, though, maybe like Mesolithic Greeks).
- A lot more mtDNA haplogroup U from really old Europeans, and mtDNA haplogroups M+N date to ~77 thousand years ago.
- Mesolithic west Europeans had blue eyes, but Neolithic Europeans had brown ones and European steppe populations "darker" than modern Europeans). ~8,000 year old Europeans had dark brown or black hair (at least two of them).
I bet 2014 will be equally exciting!
Mexicans got type 2 diabetes risk allele from Neandertals
Nature (2013) doi:10.1038/nature12828
Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico
The SIGMA Type 2 Diabetes Consortium
Performing genetic studies in multiple human populations can identify disease risk alleles that are common in one population but rare in others1, with the potential to illuminate pathophysiology, health disparities, and the population genetic origins of disease alleles. Here we analysed 9.2 million single nucleotide polymorphisms (SNPs) in each of 8,214 Mexicans and other Latin Americans: 3,848 with type 2 diabetes and 4,366 non-diabetic controls. In addition to replicating previous findings2, 3, 4, we identified a novel locus associated with type 2 diabetes at genome-wide significance spanning the solute carriers SLC16A11 and SLC16A13 (P = 3.9 × 10−13; odds ratio (OR) = 1.29). The association was stronger in younger, leaner people with type 2 diabetes, and replicated in independent samples (P = 1.1 × 10−4; OR = 1.20). The risk haplotype carries four amino acid substitutions, all in SLC16A11; it is present at ~50% frequency in Native American samples and ~10% in east Asian, but is rare in European and African samples. Analysis of an archaic genome sequence indicated that the risk haplotype introgressed into modern humans via admixture with Neanderthals. The SLC16A11 messenger RNA is expressed in liver, and V5-tagged SLC16A11 protein localizes to the endoplasmic reticulum. Expression of SLC16A11 in heterologous cells alters lipid metabolism, most notably causing an increase in intracellular triacylglycerol levels. Despite type 2 diabetes having been well studied by genome-wide association studies in other populations, analysis in Mexican and Latin American individuals identified SLC16A11 as a novel candidate gene for type 2 diabetes with a possible role in triacylglycerol metabolism.
Link
Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico
The SIGMA Type 2 Diabetes Consortium
Performing genetic studies in multiple human populations can identify disease risk alleles that are common in one population but rare in others1, with the potential to illuminate pathophysiology, health disparities, and the population genetic origins of disease alleles. Here we analysed 9.2 million single nucleotide polymorphisms (SNPs) in each of 8,214 Mexicans and other Latin Americans: 3,848 with type 2 diabetes and 4,366 non-diabetic controls. In addition to replicating previous findings2, 3, 4, we identified a novel locus associated with type 2 diabetes at genome-wide significance spanning the solute carriers SLC16A11 and SLC16A13 (P = 3.9 × 10−13; odds ratio (OR) = 1.29). The association was stronger in younger, leaner people with type 2 diabetes, and replicated in independent samples (P = 1.1 × 10−4; OR = 1.20). The risk haplotype carries four amino acid substitutions, all in SLC16A11; it is present at ~50% frequency in Native American samples and ~10% in east Asian, but is rare in European and African samples. Analysis of an archaic genome sequence indicated that the risk haplotype introgressed into modern humans via admixture with Neanderthals. The SLC16A11 messenger RNA is expressed in liver, and V5-tagged SLC16A11 protein localizes to the endoplasmic reticulum. Expression of SLC16A11 in heterologous cells alters lipid metabolism, most notably causing an increase in intracellular triacylglycerol levels. Despite type 2 diabetes having been well studied by genome-wide association studies in other populations, analysis in Mexican and Latin American individuals identified SLC16A11 as a novel candidate gene for type 2 diabetes with a possible role in triacylglycerol metabolism.
Link
December 25, 2013

December 24, 2013
Europeans = Neolithic farmers, Mesolithic hunter-gatherers and "Ancient North Eurasians" (etc.)

The Luxembourg sample is similar to the Iberian La Brana samples and the Swedish Mesolithic samples are similar to Swedish Neolithic hunter-gatherers. The LBK farmer is similar to Oetzi and a Swedish TRB farmer and to Sardinians. The authors also study the recently published Mal'ta Upper Paleolithic sample from Lake Baikal and find that it is part of an "Ancient North Eurasian" population that also admixed into West Eurasians on top of the Neolithic/Mesolithic mix.
The authors' proposed model and admixture estimates:


It seems that the estimates go all the way to "almost pure" Early European farmer ancestry but "West European Hunter-Gatherer" and "Ancient North Eurasian" ancestry isn't found unmixed in any modern populations. The model seems to agree with Raghavan et al. that Karitiana are "Mal'ta"-admixed but also finds the most basal Eurasian ancestry in the European Neolithic farmer. The authors write:
The successful model (Fig. 2A) also suggests 44 ± 10% “Basal Eurasian” admixture into the ancestors of Stuttgart: gene flow into their Near Eastern ancestors from a lineage that diverged prior to the separation of the ancestors of Loschbour and Onge. Such a scenario, while never suggested previously, is plausible given the early presence of modern humans in the Levant25, African-related tools made by modern humans in Arabia26, 27, and the geographic opportunity for continuous gene flow between the Near East and Africa28

An interesting finding is that the Luxembourg hunter-gatherer probably had blue eyes (like a Mesolithic La Brana Iberian, a paper on which seems to be in the works) but darker skin than the LBK farmer who had brown eyes but lighter skin. Raghavan et al. did not find light pigmentation in Mal'ta (but that was a very old sample), so with the exception of light eyes that seem established for Western European hunter-gatherers (and may have been "darker" in European steppe populations, but "lighter" in Bronze Age South Siberians?), the origin of depigmentation of many recent Europeans remains a mystery. Ancient DNA continues to surprise at every turn.
UPDATE (4/4/2014): a new version of the preprint.
bioRxiv doi: 10.1101/001552
Ancient human genomes suggest three ancestral populations for present-day Europeans
Iosif Lazaridis et al.
Analysis of ancient DNA can reveal historical events that are difficult to discern through study of present-day individuals. To investigate European population history around the time of the agricultural transition, we sequenced complete genomes from a ~7,500 year old early farmer from the Linearbandkeramik (LBK) culture from Stuttgart in Germany and an ~8,000 year old hunter-gatherer from the Loschbour rock shelter in Luxembourg. We also generated data from seven ~8,000 year old hunter-gatherers from Motala in Sweden. We compared these genomes and published ancient DNA to new data from 2,196 samples from 185 diverse populations to show that at least three ancestral groups contributed to present-day Europeans. The first are Ancient North Eurasians (ANE), who are more closely related to Upper Paleolithic Siberians than to any present-day population. The second are West European Hunter-Gatherers (WHG), related to the Loschbour individual, who contributed to all Europeans but not to Near Easterners. The third are Early European Farmers (EEF), related to the Stuttgart individual, who were mainly of Near Eastern origin but also harbored WHG-related ancestry. We model the deep relationships of these populations and show that about ~44% of the ancestry of EEF derived from a basal Eurasian lineage that split prior to the separation of other non-Africans.
Link
December 23, 2013
mtDNA and Y chromosomes of Tungus

Investigating the Prehistory of Tungusic Peoples of Siberia and the Amur-Ussuri Region with Complete mtDNA Genome Sequences and Y-chromosomal Markers
Ana T. Duggan et al.

Link
Recent origin of North African populations

PLoS ONE 8(11): e80293. doi:10.1371/journal.pone.0080293
Genome-Wide and Paternal Diversity Reveal a Recent Origin of Human Populations in North Africa
Karima Fadhlaoui-Zid, Marc Haber et al.
The geostrategic location of North Africa as a crossroad between three continents and as a stepping-stone outside Africa has evoked anthropological and genetic interest in this region. Numerous studies have described the genetic landscape of the human population in North Africa employing paternal, maternal, and biparental molecular markers. However, information from these markers which have different inheritance patterns has been mostly assessed independently, resulting in an incomplete description of the region. In this study, we analyze uniparental and genome-wide markers examining similarities or contrasts in the results and consequently provide a comprehensive description of the evolutionary history of North Africa populations. Our results show that both males and females in North Africa underwent a similar admixture history with slight differences in the proportions of admixture components. Consequently, genome-wide diversity show similar patterns with admixture tests suggesting North Africans are a mixture of ancestral populations related to current Africans and Eurasians with more affinity towards the out-of-Africa populations than to sub-Saharan Africans. We estimate from the paternal lineages that most North Africans emerged ~15,000 years ago during the last glacial warming and that population splits started after the desiccation of the Sahara. Although most North Africans share a common admixture history, the Tunisian Berbers show long periods of genetic isolation and appear to have diverged from surrounding populations without subsequent mixture. On the other hand, continuous gene flow from the Middle East made Egyptians genetically closer to Eurasians than to other North Africans. We show that genetic diversity of today's North Africans mostly captures patterns from migrations post Last Glacial Maximum and therefore may be insufficient to inform on the initial population of the region during the Middle Paleolithic period.
Link
December 22, 2013
Neandertals could talk
PLoS ONE 8(12): e82261. doi:10.1371/journal.pone.0082261
Micro-Biomechanics of the Kebara 2 Hyoid and Its Implications for Speech in Neanderthals
Ruggero D’Anastasio et al.
The description of a Neanderthal hyoid from Kebara Cave (Israel) in 1989 fuelled scientific debate on the evolution of speech and complex language. Gross anatomy of the Kebara 2 hyoid differs little from that of modern humans. However, whether Homo neanderthalensis could use speech or complex language remains controversial. Similarity in overall shape does not necessarily demonstrate that the Kebara 2 hyoid was used in the same way as that of Homo sapiens. The mechanical performance of whole bones is partly controlled by internal trabecular geometries, regulated by bone-remodelling in response to the forces applied. Here we show that the Neanderthal and modern human hyoids also present very similar internal architectures and micro-biomechanical behaviours. Our study incorporates detailed analysis of histology, meticulous reconstruction of musculature, and computational biomechanical analysis with models incorporating internal micro-geometry. Because internal architecture reflects the loadings to which a bone is routinely subjected, our findings are consistent with a capacity for speech in the Neanderthals.
Link
Micro-Biomechanics of the Kebara 2 Hyoid and Its Implications for Speech in Neanderthals
Ruggero D’Anastasio et al.
The description of a Neanderthal hyoid from Kebara Cave (Israel) in 1989 fuelled scientific debate on the evolution of speech and complex language. Gross anatomy of the Kebara 2 hyoid differs little from that of modern humans. However, whether Homo neanderthalensis could use speech or complex language remains controversial. Similarity in overall shape does not necessarily demonstrate that the Kebara 2 hyoid was used in the same way as that of Homo sapiens. The mechanical performance of whole bones is partly controlled by internal trabecular geometries, regulated by bone-remodelling in response to the forces applied. Here we show that the Neanderthal and modern human hyoids also present very similar internal architectures and micro-biomechanical behaviours. Our study incorporates detailed analysis of histology, meticulous reconstruction of musculature, and computational biomechanical analysis with models incorporating internal micro-geometry. Because internal architecture reflects the loadings to which a bone is routinely subjected, our findings are consistent with a capacity for speech in the Neanderthals.
Link
December 18, 2013
A Neandertal from the Altai Mountains (Prüfer et al. 2013)

Who knows what other Middle Paleolithic genomes might be in the works? My guess is that once all is said and done, the tree of Homo will fill up with "red" admixture edges, and those who argued for a single Homo lineage evolving over hundreds of thousands of years, with gene flow between regional populations, will have the upper hand.
An interesting finding is that the introgressing Neandertal (N.I.) was related to the Mezmaiskaya sample from the Caucasus rather than to the Vindija sample from Croatia or the new Altai Neandertal. It'd be great to have the genome of a bona fide "progressive" Near Eastern Neandertal.
UPDATE I (Dec. 19):
Reading the 249 pages of supplementary information is likely to reveal a lot of gems of new information.In SI 13 we see that:
We detect likely West Eurasian gene flow into the ancestors of Yoruba West Africans within the last ten thousand years, which indirectly contributed a small amount of Neandertal ancestry to Yoruba.and:
These results mean that we have not identified any sub-Saharan African sample that we are confident has no evidence of back-to-Africa migration. Our best candidate at present is the Dinka but it is possible that with a phased genome or large sample sizes we would detect evidence of non-African ancestry in this population as well.Nature (2013) doi:10.1038/nature12886
The complete genome sequence of a Neanderthal from the Altai Mountains
Kay Prüfer et al.
We present a high-quality genome sequence of a Neanderthal woman from Siberia. We show that her parents were related at the level of half-siblings and that mating among close relatives was common among her recent ancestors. We also sequenced the genome of a Neanderthal from the Caucasus to low coverage. An analysis of the relationships and population history of available archaic genomes and 25 present-day human genomes shows that several gene flow events occurred among Neanderthals, Denisovans and early modern humans, possibly including gene flow into Denisovans from an unknown archaic group. Thus, interbreeding, albeit of low magnitude, occurred among many hominin groups in the Late Pleistocene. In addition, the high-quality Neanderthal genome allows us to establish a definitive list of substitutions that became fixed in modern humans after their separation from the ancestors of Neanderthals and Denisovans.
Link
Near Eastern origin of R1a in Ashkenazi Levites
This paper is a nice cautionary tale. R1a is very common in eastern Europe and less so in the Near East. Ashkenazi Jews lived in Eastern Europe, and one group of them (Levites) had high frequency of R1a than the rest. It seemed that an eastern European patrilineage had inserted itself into the Ashkenazi Levite gene pool.
It turns out that this is not the case. The specific clade R-M582 to which Ashkenazi Levites (and other non-Levites) belong to is absent in eastern Europeans and present in non-Jewish Near Easterners, making it more likely that Jews did not pick it up from eastern Europeans, but rather from some Near Eastern population. A look at the table of frequencies suggests to me an Iranic source, but I doubt that modern populations will ever allow a full resolution of such questions.
Nature Communications 4, Article number: 2928 doi:10.1038/ncomms3928
Phylogenetic applications of whole Y-chromosome sequences and the Near Eastern origin of Ashkenazi Levites
Siiri Rootsi et al.
Previous Y-chromosome studies have demonstrated that Ashkenazi Levites, members of a paternally inherited Jewish priestly caste, display a distinctive founder event within R1a, the most prevalent Y-chromosome haplogroup in Eastern Europe. Here we report the analysis of 16 whole R1 sequences and show that a set of 19 unique nucleotide substitutions defines the Ashkenazi R1a lineage. While our survey of one of these, M582, in 2,834 R1a samples reveals its absence in 922 Eastern Europeans, we show it is present in all sampled R1a Ashkenazi Levites, as well as in 33.8% of other R1a Ashkenazi Jewish males and 5.9% of 303 R1a Near Eastern males, where it shows considerably higher diversity. Moreover, the M582 lineage also occurs at low frequencies in non-Ashkenazi Jewish populations. In contrast to the previously suggested Eastern European origin for Ashkenazi Levites, the current data are indicative of a geographic source of the Levite founder lineage in the Near East and its likely presence among pre-Diaspora Hebrews.
Link
It turns out that this is not the case. The specific clade R-M582 to which Ashkenazi Levites (and other non-Levites) belong to is absent in eastern Europeans and present in non-Jewish Near Easterners, making it more likely that Jews did not pick it up from eastern Europeans, but rather from some Near Eastern population. A look at the table of frequencies suggests to me an Iranic source, but I doubt that modern populations will ever allow a full resolution of such questions.
Nature Communications 4, Article number: 2928 doi:10.1038/ncomms3928
Phylogenetic applications of whole Y-chromosome sequences and the Near Eastern origin of Ashkenazi Levites
Siiri Rootsi et al.
Previous Y-chromosome studies have demonstrated that Ashkenazi Levites, members of a paternally inherited Jewish priestly caste, display a distinctive founder event within R1a, the most prevalent Y-chromosome haplogroup in Eastern Europe. Here we report the analysis of 16 whole R1 sequences and show that a set of 19 unique nucleotide substitutions defines the Ashkenazi R1a lineage. While our survey of one of these, M582, in 2,834 R1a samples reveals its absence in 922 Eastern Europeans, we show it is present in all sampled R1a Ashkenazi Levites, as well as in 33.8% of other R1a Ashkenazi Jewish males and 5.9% of 303 R1a Near Eastern males, where it shows considerably higher diversity. Moreover, the M582 lineage also occurs at low frequencies in non-Ashkenazi Jewish populations. In contrast to the previously suggested Eastern European origin for Ashkenazi Levites, the current data are indicative of a geographic source of the Levite founder lineage in the Near East and its likely presence among pre-Diaspora Hebrews.
Link
December 17, 2013
Reconstruction of 5,500-year old "Stonehenge Man"
 I don't see any mention of DNA in the article The face of prehistoric Britain: Forensic scientist uses Neolithic man's 5,500-year-old skull to create lifelike image as part of new £27m Stonehenge centre, so it's not clear whether the pigmentation attributed to "Stonehenge Man" is the artist's imagination or based on solid evidence.
I don't see any mention of DNA in the article The face of prehistoric Britain: Forensic scientist uses Neolithic man's 5,500-year-old skull to create lifelike image as part of new £27m Stonehenge centre, so it's not clear whether the pigmentation attributed to "Stonehenge Man" is the artist's imagination or based on solid evidence.From the article:
He is the star attraction of Stonehenge's new £27million modern visitor centre that has taken decades to produce.
A Neolithic man has been brought to life after the most advanced forensic reconstruction of a face, based on a 5,500-year-old skeleton buried in a long barrow 1.5 miles from Stonehenge.
The new face of the model, which has been carefully reconstructed to show people what life was like
December 15, 2013
Arabian origin of the Upper Paleolithic in the Levant
This is a very useful review of research on the origin of the Upper Paleolithic (Emiran) in the Levant, arguing against a recent (c. 50kya ) African origin and in favor of an Arabian one. The argument is mainly archaeological, although it is informed by genetic evidence. From the chapter:
A century of research into the origins of the Upper Palaeolithic in the Levant
Anthony E. Marks and Jeffrey I. Rose
Link
After a century of research, the origins of the Levantine UP still remain an enigma. At this point, at least one thing is clear: the Emiran has no African progenitor. As such, there is a disconnect between the archaeological database and the Replacement paradigm, which necessitates that the earliest Levantine Upper Paleolithic must have come fully developed from northeast Africa. The Replacement model should have been a parsimonious prism through which to view the transition from the MP to the UP in the Levant. It was not.The recent acceptance of: (i) a slower autosomal mutation rate, and (ii) evidence for interbreeding with Neandertals largely predating the c. 50kya mark, and (iii) coalescence of Eurasian mtDNA haplogroup N well before that time, have all but killed, in my opinion the idea of a 50kya spread of modern humans from Africa. Modern humans must have lived in Eurasia much earlier than that time, and what remains is to figure out how much earlier.
A century of research into the origins of the Upper Palaeolithic in the Levant
Anthony E. Marks and Jeffrey I. Rose
Link
December 14, 2013
Ancient mtDNA from Rössen culture in Wittmar, Germany
Archaeological and Anthropological Sciences December 2013
Ancient DNA insights from the Middle Neolithic in Germany
Esther J. Lee et al.
Genetic studies of Neolithic groups in central Europe have provided insights into the demographic processes that have occurred during the initial transition to agriculture as well as in later Neolithic contexts. While distinct genetic patterns between indigenous hunter-gatherers and Neolithic farmers in Europe have been observed, it is still under discussion how the genetic diversity changed during the 5,000-year span of the Neolithic period. In order to investigate genetic patterns after the earliest farming communities, we carried out an ancient mitochondrial DNA (mtDNA) analysis of 34 individuals from Wittmar, Germany representing three different Neolithic farming groups (ca. 5,200–4,300 cal bc) including Rössen societies. Ancient DNA analysis was successful for six individuals associated with the Middle Neolithic Rössen and observed haplotypes were assigned to mtDNA haplogroups H5, HV0, U5, and K. Our results offer perspectives on the genetic composition of individuals associated with the Rössen culture at Wittmar and permit insights into genetic landscapes in central Europe at a time when regional groups first emerged during the Middle Neolithic.
Link
Ancient DNA insights from the Middle Neolithic in Germany
Esther J. Lee et al.
Genetic studies of Neolithic groups in central Europe have provided insights into the demographic processes that have occurred during the initial transition to agriculture as well as in later Neolithic contexts. While distinct genetic patterns between indigenous hunter-gatherers and Neolithic farmers in Europe have been observed, it is still under discussion how the genetic diversity changed during the 5,000-year span of the Neolithic period. In order to investigate genetic patterns after the earliest farming communities, we carried out an ancient mitochondrial DNA (mtDNA) analysis of 34 individuals from Wittmar, Germany representing three different Neolithic farming groups (ca. 5,200–4,300 cal bc) including Rössen societies. Ancient DNA analysis was successful for six individuals associated with the Middle Neolithic Rössen and observed haplotypes were assigned to mtDNA haplogroups H5, HV0, U5, and K. Our results offer perspectives on the genetic composition of individuals associated with the Rössen culture at Wittmar and permit insights into genetic landscapes in central Europe at a time when regional groups first emerged during the Middle Neolithic.
Link
December 12, 2013
No evidence for selection since admixture in sample of 29,141 African Americans
arXiv:1312.2675 [q-bio.PE]
Genome-wide scan of 29,141 African Americans finds no evidence of selection since admixture
Gaurav Bhatia et al.
We scanned through the genomes of 29,141 African Americans, searching for loci where the average proportion of African ancestry deviates significantly from the genome-wide average. We failed to find any genome-wide significant deviations, and conclude that any selection in African Americans since admixture is sufficiently weak that it falls below the threshold of our power to detect it using a large sample size. These results stand in contrast to the findings of a recent study of selection in African Americans. That study, which had 15 times fewer samples, reported six loci with significant deviations. We show that the discrepancy is likely due to insufficient correction for multiple hypothesis testing in the previous study. The same study reported 14 loci that showed greater population differentiation between African Americans and Nigerian Yoruba than would be expected in the absence of natural selection. Four such loci were previously shown to be genome-wide significant and likely to be affected by selection, but we show that most of the 10 additional loci are likely to be false positives. Additionally, the most parsimonious explanation for the loci that have significant evidence of unusual differentiation in frequency between Nigerians and Africans Americans is selection in Africa prior to their forced migration to the Americas.
Link
Genome-wide scan of 29,141 African Americans finds no evidence of selection since admixture
Gaurav Bhatia et al.
We scanned through the genomes of 29,141 African Americans, searching for loci where the average proportion of African ancestry deviates significantly from the genome-wide average. We failed to find any genome-wide significant deviations, and conclude that any selection in African Americans since admixture is sufficiently weak that it falls below the threshold of our power to detect it using a large sample size. These results stand in contrast to the findings of a recent study of selection in African Americans. That study, which had 15 times fewer samples, reported six loci with significant deviations. We show that the discrepancy is likely due to insufficient correction for multiple hypothesis testing in the previous study. The same study reported 14 loci that showed greater population differentiation between African Americans and Nigerian Yoruba than would be expected in the absence of natural selection. Four such loci were previously shown to be genome-wide significant and likely to be affected by selection, but we show that most of the 10 additional loci are likely to be false positives. Additionally, the most parsimonious explanation for the loci that have significant evidence of unusual differentiation in frequency between Nigerians and Africans Americans is selection in Africa prior to their forced migration to the Americas.
Link
Ancient DNA meeting talks audio
Some of the audio from talks of this meeting that I posted about before have been posted. I don't think I can listen to all of them, but feel free to post any interesting "nuggets" of information in the comments.
December 06, 2013
Merovingian mtDNA
From the paper:
Ancient DNA and kinship analysis of human remains deposited in Merovingian necropolis sarcophagi (Jau Dignac et Loirac, France, 7th–8th century AD)
M.F. Deguilloux et al.
The analysis of ancient DNA recovered from archaeological remains can be used to reconstruct kinship among the occupants of a necropolis and provide a more detailed portrait of the community considered. Such palaeogenetic analyses have been conducted on sarcophagi excavated from the Merovingian necropolis in Jau-Dignac et Loirac (7th–8th century AD, Aquitaine, southwest France). The genetic study consisted of the analysis of mitochondrial DNA and nuclear STRs (Short Tandem Repeats) from nine skeletons deposited in three grouped sarcophagi. Only data concerning the mitochondrial genomes could be obtained, and six different mitochondrial lineages were retrieved from eight samples. Our analyses permitted a high confidence characterisation of maternal relationships between individuals deposited in the same sepulchre. These results are important and novel for the period and region and argue that individuals were grouped inside sarcophagi according to relationship criteria. The presence of perinatal remains in one sarcophagus was particularly striking because access to this type of funerary structure during this period was generally reserved for older children. Moreover, we demonstrated genetically that the perinatal remains were not related maternally to two women found in the same sarcophagus (whereas the maternal relationship between the two young women could be determined), and we proposed different possible explanations for this unexpected observation. Overall, archaeological, anthropological and genetic data suggest that the Jau-Dignac et Loirac necropolis groups together the closely and distantly related members of a High Middle Ages familia. Our ancient DNA analyses note the important contribution of palaeogenetic analyses to archaeological kinship studies.
Link
Our approach clearly identified six different mitochondrial lineages (corresponding to five distinct haplogroups: J, H, K, X2 and W) among eight human remains, indicating noticeable mitochondrial diversity. During this period, the site might have been the cemetery for a social group with significant genetic diversity.Journal of Archaeological Science Volume 41, January 2014, Pages 399–405
Ancient DNA and kinship analysis of human remains deposited in Merovingian necropolis sarcophagi (Jau Dignac et Loirac, France, 7th–8th century AD)
M.F. Deguilloux et al.
The analysis of ancient DNA recovered from archaeological remains can be used to reconstruct kinship among the occupants of a necropolis and provide a more detailed portrait of the community considered. Such palaeogenetic analyses have been conducted on sarcophagi excavated from the Merovingian necropolis in Jau-Dignac et Loirac (7th–8th century AD, Aquitaine, southwest France). The genetic study consisted of the analysis of mitochondrial DNA and nuclear STRs (Short Tandem Repeats) from nine skeletons deposited in three grouped sarcophagi. Only data concerning the mitochondrial genomes could be obtained, and six different mitochondrial lineages were retrieved from eight samples. Our analyses permitted a high confidence characterisation of maternal relationships between individuals deposited in the same sepulchre. These results are important and novel for the period and region and argue that individuals were grouped inside sarcophagi according to relationship criteria. The presence of perinatal remains in one sarcophagus was particularly striking because access to this type of funerary structure during this period was generally reserved for older children. Moreover, we demonstrated genetically that the perinatal remains were not related maternally to two women found in the same sarcophagus (whereas the maternal relationship between the two young women could be determined), and we proposed different possible explanations for this unexpected observation. Overall, archaeological, anthropological and genetic data suggest that the Jau-Dignac et Loirac necropolis groups together the closely and distantly related members of a High Middle Ages familia. Our ancient DNA analyses note the important contribution of palaeogenetic analyses to archaeological kinship studies.
Link
December 05, 2013
Early 7th millennium BC Initial Neolithic in Franchthi Cave
Antiquity Volume: 87 Number: 338 Page: 1001–1015
Early seventh-millennium AMS dates from domestic seeds in the Initial Neolithic at Franchthi Cave (Argolid, Greece)
Catherine Perlès1, Anita Quiles2 and Hélène Valladas2
When, and by what route, did farming first reach Europe? A terrestrial model might envisage a gradual advance around the northern fringes of the Aegean, reaching Thrace and Macedonia before continuing southwards to Thessaly and the Peloponnese. New dates from Franchthi Cave in southern Greece, reported here, cast doubt on such a model, indicating that cereal cultivation, involving newly introduced crop species, began during the first half of the seventh millennium BC. This is earlier than in northern Greece and several centuries earlier than in Bulgaria, and suggests that farming spread to south-eastern Europe by a number of different routes, including potentially a maritime, island-hopping connection across the Aegean Sea. The results also illustrate the continuing importance of key sites such as Franchthi to our understanding of the European Neolithic transition, and the additional insights that can emerge from the application of new dating projects to these sites.
Link
Early seventh-millennium AMS dates from domestic seeds in the Initial Neolithic at Franchthi Cave (Argolid, Greece)
Catherine Perlès1, Anita Quiles2 and Hélène Valladas2
When, and by what route, did farming first reach Europe? A terrestrial model might envisage a gradual advance around the northern fringes of the Aegean, reaching Thrace and Macedonia before continuing southwards to Thessaly and the Peloponnese. New dates from Franchthi Cave in southern Greece, reported here, cast doubt on such a model, indicating that cereal cultivation, involving newly introduced crop species, began during the first half of the seventh millennium BC. This is earlier than in northern Greece and several centuries earlier than in Bulgaria, and suggests that farming spread to south-eastern Europe by a number of different routes, including potentially a maritime, island-hopping connection across the Aegean Sea. The results also illustrate the continuing importance of key sites such as Franchthi to our understanding of the European Neolithic transition, and the additional insights that can emerge from the application of new dating projects to these sites.
Link
December 04, 2013
400 thousand year old human mtDNA from Sima de los Huesos

A new paper in Nature reports their success, and overnight increases by an order of magnitude the time depth for which we now have human mtDNA from what is commonly designated as Homo heidelbergensis, from right in the middle of the Middle Pleistocene. Obviously, this opens new vistas for archaeogenetic research, making it possible to directly look at early pre-sapiens forms of humans, and not only on their final forms prior to their replacement, the Neandertals and Denisovans.
The most impressive aspect of the new paper is most likely the technical challenges that the researchers must've overcome to achieve this result. The cave bear DNA showed that this was possible, but human DNA adds an additional complication in the form of contamination by a closely related species, us.
But, the new evolutionary result which will interest those of us not interested in the minutiae of biomolecules will no doubt be the fact that the Sima hominin's mtDNA formed a clade with the much more recent Denisova girl.
Until now, we knew that Neandertal mtDNA grouped together and so did modern human mtDNA. The two groups shared a Middle Pleistocene common ancestor and a much more distant common ancestor (~1 million years) with the mtDNA found in Denisova. The new Sima specimen shares descent from Denisova. This is important because it shows that whatever archaic human population the Denisovan mtDNA belonged to also extended to western Europe. And, surprisingly, the Sima specimen did not group with Neandertals, as might be expected because of the incipient Neanderthaloid morphology of the Sima hominins which has been a matter of controversy as it pushes back the evolutionary lineage of H. neandertalensis deeper into the Middle Pleistocene that some researchers accept.
Before this paper, it was believed that H. heidelbergensis evolved somewhere (perhaps Near East or Africa), a subset of it evolved to H. sapiens in Africa, and a different subset evolved in Eurasia, leading up to H. neandertalensis in the west, and unknown forms in the east, of which the Denisova girl was a matrilineal descendant. The next question is: when did Neandertals and Neandertal mtDNA appear in Europe?
It can now be hoped that such questions will be answered directly. The Sima individual studied in this paper is not some frozen specimen from the Arctic, preserved by a freak accident in pristine form for hundreds of thousands of years, but a person who lived in Southwestern Europe. I am fairly sure that this won't be the last really old human we see a paper about in the coming years. Human mtDNA used to present a simple picture at the time of the discovery of African mitochondrial Eve: the deepest splits were in Africa and Eurasians belonged to a subset of African variation. But, as more and more archaic Eurasian mtDNA is sampled, it now appears that modern human mtDNA is a subset of world human mtDNA whose deepest splits are in Eurasia, and the next deepest splits are in Africa. Obviously, this may be a consequence of the fact that archaic human mtDNA has only been sampled from Eurasia, for factors relating to DNA preservation. But, it is nonetheless interesting to wonder where on the tree the mtDNA of archaic Africans would fall.
Nature (2013) doi:10.1038/nature12788
A mitochondrial genome sequence of a hominin from Sima de los Huesos
Matthias Meyer et al.
Excavations of a complex of caves in the Sierra de Atapuerca in northern Spain have unearthed hominin fossils that range in age from the early Pleistocene to the Holocene1. One of these sites, the ‘Sima de los Huesos’ (‘pit of bones’), has yielded the world’s largest assemblage of Middle Pleistocene hominin fossils2, 3, consisting of at least 28 individuals4 dated to over 300,000 years ago5. The skeletal remains share a number of morphological features with fossils classified as Homo heidelbergensis and also display distinct Neanderthal-derived traits6, 7, 8. Here we determine an almost complete mitochondrial genome sequence of a hominin from Sima de los Huesos and show that it is closely related to the lineage leading to mitochondrial genomes of Denisovans9, 10, an eastern Eurasian sister group to Neanderthals. Our results pave the way for DNA research on hominins from the Middle Pleistocene.
Link
November 28, 2013
Iberian Neolithic farmer DNA
A currently not available preprint that has important implications about the Neolithic of Europe.
A late Neolithic Iberian farmer exhibits genetic affinity to Neolithic Scandinavian farmers and a Bronze Age central European farmer
Sverrisdóttir, Oddný Ósk et al.
The spread of farming, the neolithisation process, swept over Europe after the advent of the farming lifestyle in the near east approximately 11,000 years ago. However the mode of transmission and its impact on the demographic patterns of Europe remains largely unknown. In this study we obtained : 66,476,944 bp of genomic DNA from the remains of a 4000 year old Neolithic farmer from the site of El Portalón, 15 km east of Burgos, Spain. We compared the genomic signature of this individual to modern-day populations as well as the few Neolithic individuals that has produced large-scale autosomal data. The Neolithic Portalón individual is genetically most similar to southern Europeans, similar to a Scandinavian Neolithic farmer and the Tyrolean Iceman. In contrast, the Neolithic Portalón individual displays little affinity to two Mesolithic samples from the near-by area, La Brana, demonstrating a distinct change in population history between 7,000 and 4,000 years ago for the northern Iberian Peninsula.
Link
A late Neolithic Iberian farmer exhibits genetic affinity to Neolithic Scandinavian farmers and a Bronze Age central European farmer
Sverrisdóttir, Oddný Ósk et al.
The spread of farming, the neolithisation process, swept over Europe after the advent of the farming lifestyle in the near east approximately 11,000 years ago. However the mode of transmission and its impact on the demographic patterns of Europe remains largely unknown. In this study we obtained : 66,476,944 bp of genomic DNA from the remains of a 4000 year old Neolithic farmer from the site of El Portalón, 15 km east of Burgos, Spain. We compared the genomic signature of this individual to modern-day populations as well as the few Neolithic individuals that has produced large-scale autosomal data. The Neolithic Portalón individual is genetically most similar to southern Europeans, similar to a Scandinavian Neolithic farmer and the Tyrolean Iceman. In contrast, the Neolithic Portalón individual displays little affinity to two Mesolithic samples from the near-by area, La Brana, demonstrating a distinct change in population history between 7,000 and 4,000 years ago for the northern Iberian Peninsula.
Link
November 26, 2013
One to three men fathered most western Europeans?
It may sound far-fetched but it's certainly possible. After all, no R1b has been found in Europe before a Bell Beaker site from the 3rd millennium BC and today many Europeans (most in western Europe) belong to this haplogroup. As more Y chromosomes are sampled from ancient Europe, it will become clear if the R1b frequency actually shot from non-existence to ubiquity over a short span of time, and the Y chromosomes after the transition will be practically clones of each other.
Investigative Genetics 2013, 4:25 doi:10.1186/2041-2223-4-25
Modeling the contrasting Neolithic male lineage expansions in Europe and Africa
Michael J Sikora et al.
Abstract (provisional)
Background
Patterns of genetic variation in a population carry information about the prehistory of the population, and for the human Y chromosome an especially informative phylogenetic tree has previously been constructed from fully-sequenced chromosomes. This revealed contrasting bifurcating and starlike phylogenies for the major lineages associated with the Neolithic expansions in sub-Saharan Africa and Western Europe, respectively.
Results
We used coalescent simulations to investigate the range of demographic models most likely to produce the phylogenetic structures observed in Africa and Europe, assessing the starting and ending genetic effective population sizes, duration of the expansion, and time when expansion ended. The best-fitting models in Africa and Europe are very different. In Africa, the expansion took about 12 thousand years, ending very recently; it started from approximately 40 men and numbers expanded approximately 50-fold. In Europe, the expansion was much more rapid, taking only a few generations and occurring as soon as the major R1b lineage entered Europe; it started from just one to three men, whose numbers expanded more than a thousandfold.
Conclusions
Although highly simplified, the demographic model we have used captures key elements of the differences between the male Neolithic expansions in Africa and Europe, and is consistent with archaeological findings.
Link
Investigative Genetics 2013, 4:25 doi:10.1186/2041-2223-4-25
Modeling the contrasting Neolithic male lineage expansions in Europe and Africa
Michael J Sikora et al.
Abstract (provisional)
Background
Patterns of genetic variation in a population carry information about the prehistory of the population, and for the human Y chromosome an especially informative phylogenetic tree has previously been constructed from fully-sequenced chromosomes. This revealed contrasting bifurcating and starlike phylogenies for the major lineages associated with the Neolithic expansions in sub-Saharan Africa and Western Europe, respectively.
Results
We used coalescent simulations to investigate the range of demographic models most likely to produce the phylogenetic structures observed in Africa and Europe, assessing the starting and ending genetic effective population sizes, duration of the expansion, and time when expansion ended. The best-fitting models in Africa and Europe are very different. In Africa, the expansion took about 12 thousand years, ending very recently; it started from approximately 40 men and numbers expanded approximately 50-fold. In Europe, the expansion was much more rapid, taking only a few generations and occurring as soon as the major R1b lineage entered Europe; it started from just one to three men, whose numbers expanded more than a thousandfold.
Conclusions
Although highly simplified, the demographic model we have used captures key elements of the differences between the male Neolithic expansions in Africa and Europe, and is consistent with archaeological findings.
Link
A priori Y chromosome phylogeny from sequencing data
A cool new paper by a team of citizen scientists. The most important new piece of evidence is the joining together of haplogroup M (Papuans) with P in a new MP internal node. Your guess is as good as mine as to whether this MP may have come from, as his descendants are presently spread from the Atlantic via Siberia to the Amazon and all the way to New Guinea. The Mal'ta boy belonged to haplogroup R.
The other interesting discovery is of one Telugu man from India who shares mutations with haplogroups N and O but belongs to neither N nor O, so this defines a new "X" clade in the phylogeny. I am wondering if this could perhaps be called NO0 instead, similar to the way that more basal clades of the entire phylogeny were called A0, A00, and so on? Terminology is tricky...

I am aware of a few commercial ventures to resequence Y chromosomes, and I'm pretty sure that citizen scientists will soon not only be able to re-analyze data such as those from the 1000 Genomes Project, but will be able to generate data of their own.
bioRxiv doi: 10.1101/000802
Generation of high-resolution a priori Y-chromosome phylogenies using “next-generation” sequencing data
Gregory R Magoon et al.
An approach for generating high-resolution a priori maximum parsimony Y-chromosome (“chrY”) phylogenies based on SNP and small INDEL variant data from massively-parallel short-read (“next-generation”) sequencing data is described; the tree-generation methodology produces annotations localizing mutations to individual branches of the tree, along with indications of mutation placement uncertainty in cases for which "no-calls" (through lack of mapped reads or otherwise) at particular site precludes a precise placement of the mutation. The approach leverages careful variant site filtering and a novel iterative reweighting procedure to generate high-accuracy trees while considering variants in regions of chrY that had previously been excluded from analyses based on short-read sequencing data. It is argued that the proposed approach is also superior to previous region-based filtering approaches in that it adapts to the quality of the underlying data and will automatically allow the scope of sites considered to expand as the underlying data quality (e.g. through longer read lengths) improves. Key related issues, including calling of genotypes for the hemizygous chrY, reliability of variant results, read mismappings and "heterozygous" genotype calls, and the mutational stability of different variants are discussed and taken into account. The methodology is demonstrated through application to a dataset consisting of 1292 male samples from diverse populations and haplogroups, with the majority coming from low-coverage sequencing by the 1000 Genomes Project. Application of the tree-generation approach to these data produces a tree involving over 120,000 chrY variant sites (about 45,000 sites if “singletons” are excluded). The utility of this approach in refining the Y-chromosome phylogenetic tree is demonstrated by examining results for several haplogroups. The results indicate a number of new branches on the Y-chromosome phylogenetic tree, many of them subdividing known branches, but also including some that inform the presence of additional levels along the “trunk” of the tree. Finally, opportunities for extensions of this phylogenetic analysis approach to other types of genetic data are examined.
Link
The other interesting discovery is of one Telugu man from India who shares mutations with haplogroups N and O but belongs to neither N nor O, so this defines a new "X" clade in the phylogeny. I am wondering if this could perhaps be called NO0 instead, similar to the way that more basal clades of the entire phylogeny were called A0, A00, and so on? Terminology is tricky...

I am aware of a few commercial ventures to resequence Y chromosomes, and I'm pretty sure that citizen scientists will soon not only be able to re-analyze data such as those from the 1000 Genomes Project, but will be able to generate data of their own.
bioRxiv doi: 10.1101/000802
Generation of high-resolution a priori Y-chromosome phylogenies using “next-generation” sequencing data
Gregory R Magoon et al.
An approach for generating high-resolution a priori maximum parsimony Y-chromosome (“chrY”) phylogenies based on SNP and small INDEL variant data from massively-parallel short-read (“next-generation”) sequencing data is described; the tree-generation methodology produces annotations localizing mutations to individual branches of the tree, along with indications of mutation placement uncertainty in cases for which "no-calls" (through lack of mapped reads or otherwise) at particular site precludes a precise placement of the mutation. The approach leverages careful variant site filtering and a novel iterative reweighting procedure to generate high-accuracy trees while considering variants in regions of chrY that had previously been excluded from analyses based on short-read sequencing data. It is argued that the proposed approach is also superior to previous region-based filtering approaches in that it adapts to the quality of the underlying data and will automatically allow the scope of sites considered to expand as the underlying data quality (e.g. through longer read lengths) improves. Key related issues, including calling of genotypes for the hemizygous chrY, reliability of variant results, read mismappings and "heterozygous" genotype calls, and the mutational stability of different variants are discussed and taken into account. The methodology is demonstrated through application to a dataset consisting of 1292 male samples from diverse populations and haplogroups, with the majority coming from low-coverage sequencing by the 1000 Genomes Project. Application of the tree-generation approach to these data produces a tree involving over 120,000 chrY variant sites (about 45,000 sites if “singletons” are excluded). The utility of this approach in refining the Y-chromosome phylogenetic tree is demonstrated by examining results for several haplogroups. The results indicate a number of new branches on the Y-chromosome phylogenetic tree, many of them subdividing known branches, but also including some that inform the presence of additional levels along the “trunk” of the tree. Finally, opportunities for extensions of this phylogenetic analysis approach to other types of genetic data are examined.
Link
November 20, 2013
Ancient DNA from Upper Paleolithic Lake Baikal (Mal'ta and Afontova Gora)
The study I mentioned in a previous post has now been made available in Nature. Two Upper Paleolithic Siberians (24-17kya) have been sequenced at low coverage. The better quality (and older) Mal'ta (MA-1) sample belongs to Y-haplogroup R and mtDNA haplogroup U, and the younger (but poorer quality) Afontova Gora (AG-2) sample appears to be related to it.
 Most interestingly, there is evidence for gene flow between the MA-1 sample and Native Americans, which makes sense as these are Siberians of the period leading up to the initial colonization of the Americas. The interesting thing is that MA-1 does not appear to be East Eurasian, as proven by the test D(Papuan, Han; Sardinian, MA-1) which is non-significant, so MA-1 is not more closely related to Han than to Papuans (which is true for modern native Americans). So, it seems that the gene flow between MA-1 and Native Americans was towards Native Americans from MA-1 and not vice versa.
Most interestingly, there is evidence for gene flow between the MA-1 sample and Native Americans, which makes sense as these are Siberians of the period leading up to the initial colonization of the Americas. The interesting thing is that MA-1 does not appear to be East Eurasian, as proven by the test D(Papuan, Han; Sardinian, MA-1) which is non-significant, so MA-1 is not more closely related to Han than to Papuans (which is true for modern native Americans). So, it seems that the gene flow between MA-1 and Native Americans was towards Native Americans from MA-1 and not vice versa.
It is fascinating that such a sample could be found so far east at so early a time. Both Y-chromosome R and mtDNA haplogroup U are very rare east of Lake Baikal which has been considered a limit of west Eurasian influence into east Eurasia. And, indeed, both these haplogroups are absent in Native Americans, so it is not yet clear how Native Americans (who belong to Y-chromosome haplogroups Q and C and mtDNA haplogroups A, B, C, D, X) are related to these Paleolithic Siberians. The obvious candidate for this relationship is Y-chromosome haplogroup P (the ancestor of Q and R). So, perhaps Q-bearing relatives of the R-bearing Mal'ta population settled the Americas.
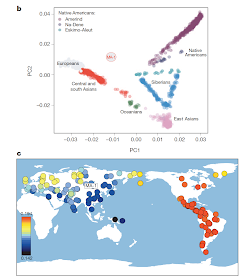 In any case, this is an extremely important sample, as its position in "no man's land" in the PCA plot (left) demonstrates, between Europeans and native Americans but close to no modern population.
In any case, this is an extremely important sample, as its position in "no man's land" in the PCA plot (left) demonstrates, between Europeans and native Americans but close to no modern population.
Its closest present-day relatives are indicated in (c), with Native Americans (red) being the closest, and a scattering of boreal populations from the Atlantic to the Pacific (but not in the vicinity of Lake Baikal) next in line (yellow).
This distribution clearly related to the evidence for admixture in Europe adduced in two other recent papers, although the question of who went where and when remains to be resolved. Was MA-1 part of an intrusive western population encroaching on east Eurasians? Or did MA-1 lookalikes arrive as first settlers in empty territory, later ceding this space to east Eurasians from, perhaps, China? Did the two mix in Siberia or did they arrive in the Americas in separate migrations and mix there? And, how does this all relate to events in Europe in the far west?
UPDATE: Razib makes an excellent point:
Nature (2013) doi:10.1038/nature12736
Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans
Maanasa Raghavan, Pontus Skoglund et al.
The origins of the First Americans remain contentious. Although Native Americans seem to be genetically most closely related to east Asians1, 2, 3, there is no consensus with regard to which specific Old World populations they are closest to4, 5, 6, 7, 8. Here we sequence the draft genome of an approximately 24,000-year-old individual (MA-1), from Mal’ta in south-central Siberia9, to an average depth of 1×. To our knowledge this is the oldest anatomically modern human genome reported to date. The MA-1 mitochondrial genome belongs to haplogroup U, which has also been found at high frequency among Upper Palaeolithic and Mesolithic European hunter-gatherers10, 11, 12, and the Y chromosome of MA-1 is basal to modern-day western Eurasians and near the root of most Native American lineages5. Similarly, we find autosomal evidence that MA-1 is basal to modern-day western Eurasians and genetically closely related to modern-day Native Americans, with no close affinity to east Asians. This suggests that populations related to contemporary western Eurasians had a more north-easterly distribution 24,000 years ago than commonly thought. Furthermore, we estimate that 14 to 38% of Native American ancestry may originate through gene flow from this ancient population. This is likely to have occurred after the divergence of Native American ancestors from east Asian ancestors, but before the diversification of Native American populations in the New World. Gene flow from the MA-1 lineage into Native American ancestors could explain why several crania from the First Americans have been reported as bearing morphological characteristics that do not resemble those of east Asians2, 13. Sequencing of another south-central Siberian, Afontova Gora-2 dating to approximately 17,000 years ago14, revealed similar autosomal genetic signatures as MA-1, suggesting that the region was continuously occupied by humans throughout the Last Glacial Maximum. Our findings reveal that western Eurasian genetic signatures in modern-day Native Americans derive not only from post-Columbian admixture, as commonly thought, but also from a mixed ancestry of the First Americans.
Link

It is fascinating that such a sample could be found so far east at so early a time. Both Y-chromosome R and mtDNA haplogroup U are very rare east of Lake Baikal which has been considered a limit of west Eurasian influence into east Eurasia. And, indeed, both these haplogroups are absent in Native Americans, so it is not yet clear how Native Americans (who belong to Y-chromosome haplogroups Q and C and mtDNA haplogroups A, B, C, D, X) are related to these Paleolithic Siberians. The obvious candidate for this relationship is Y-chromosome haplogroup P (the ancestor of Q and R). So, perhaps Q-bearing relatives of the R-bearing Mal'ta population settled the Americas.

Its closest present-day relatives are indicated in (c), with Native Americans (red) being the closest, and a scattering of boreal populations from the Atlantic to the Pacific (but not in the vicinity of Lake Baikal) next in line (yellow).
This distribution clearly related to the evidence for admixture in Europe adduced in two other recent papers, although the question of who went where and when remains to be resolved. Was MA-1 part of an intrusive western population encroaching on east Eurasians? Or did MA-1 lookalikes arrive as first settlers in empty territory, later ceding this space to east Eurasians from, perhaps, China? Did the two mix in Siberia or did they arrive in the Americas in separate migrations and mix there? And, how does this all relate to events in Europe in the far west?
UPDATE: Razib makes an excellent point:
Also, can we now finally bury the debate when east and west Eurasians diverged? Obviously it can’t have been that recent if a >20,000 year old individual had closer affinity to western populations.We already knew that Tianyuan was more Asian than European, so I think west Eurasians diverged from the rest >40 thousand years ago. But, Tianyuan was so early that its precise relationships to different Asian groups could not be determined. So, I'd say it's a good guess that east-west split off before 40 thousand years in Eurasia.
Nature (2013) doi:10.1038/nature12736
Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans
Maanasa Raghavan, Pontus Skoglund et al.
The origins of the First Americans remain contentious. Although Native Americans seem to be genetically most closely related to east Asians1, 2, 3, there is no consensus with regard to which specific Old World populations they are closest to4, 5, 6, 7, 8. Here we sequence the draft genome of an approximately 24,000-year-old individual (MA-1), from Mal’ta in south-central Siberia9, to an average depth of 1×. To our knowledge this is the oldest anatomically modern human genome reported to date. The MA-1 mitochondrial genome belongs to haplogroup U, which has also been found at high frequency among Upper Palaeolithic and Mesolithic European hunter-gatherers10, 11, 12, and the Y chromosome of MA-1 is basal to modern-day western Eurasians and near the root of most Native American lineages5. Similarly, we find autosomal evidence that MA-1 is basal to modern-day western Eurasians and genetically closely related to modern-day Native Americans, with no close affinity to east Asians. This suggests that populations related to contemporary western Eurasians had a more north-easterly distribution 24,000 years ago than commonly thought. Furthermore, we estimate that 14 to 38% of Native American ancestry may originate through gene flow from this ancient population. This is likely to have occurred after the divergence of Native American ancestors from east Asian ancestors, but before the diversification of Native American populations in the New World. Gene flow from the MA-1 lineage into Native American ancestors could explain why several crania from the First Americans have been reported as bearing morphological characteristics that do not resemble those of east Asians2, 13. Sequencing of another south-central Siberian, Afontova Gora-2 dating to approximately 17,000 years ago14, revealed similar autosomal genetic signatures as MA-1, suggesting that the region was continuously occupied by humans throughout the Last Glacial Maximum. Our findings reveal that western Eurasian genetic signatures in modern-day Native Americans derive not only from post-Columbian admixture, as commonly thought, but also from a mixed ancestry of the First Americans.
Link
November 18, 2013
Royal Society Ancient DNA meeting
Ancient DNA: the first three decades
Ancient DNA: applications in human evolutionary history (this also has abstracts)
Program for both days (pdf). I've seen some tweets from it using the #ancientDNA tag.
Ancient DNA: applications in human evolutionary history (this also has abstracts)
Program for both days (pdf). I've seen some tweets from it using the #ancientDNA tag.
November 15, 2013
Music and population structure (Brown et al. 2013)
Proceedings of the Royal Society B doi: 10.1098/rspb.2013.2072
Correlations in the population structure of music, genes and language
Steven Brown et al.
We present, to our knowledge, the first quantitative evidence that music and genes may have coevolved by demonstrating significant correlations between traditional group-level folk songs and mitochondrial DNA variation among nine indigenous populations of Taiwan. These correlations were of comparable magnitude to those between language and genes for the same populations, although music and language were not significantly correlated with one another. An examination of population structure for genetics showed stronger parallels to music than to language. Overall, the results suggest that music might have a sufficient time-depth to retrace ancient population movements and, additionally, that it might be capturing different aspects of population history than language. Music may therefore have the potential to serve as a novel marker of human migrations to complement genes, language and other markers.
Link
Correlations in the population structure of music, genes and language
Steven Brown et al.
We present, to our knowledge, the first quantitative evidence that music and genes may have coevolved by demonstrating significant correlations between traditional group-level folk songs and mitochondrial DNA variation among nine indigenous populations of Taiwan. These correlations were of comparable magnitude to those between language and genes for the same populations, although music and language were not significantly correlated with one another. An examination of population structure for genetics showed stronger parallels to music than to language. Overall, the results suggest that music might have a sufficient time-depth to retrace ancient population movements and, additionally, that it might be capturing different aspects of population history than language. Music may therefore have the potential to serve as a novel marker of human migrations to complement genes, language and other markers.
Link
Population history of the Caribbean

Reconstructing the Population Genetic History of the Caribbean
Andrés Moreno-Estrada et al.
The Caribbean basin is home to some of the most complex interactions in recent history among previously diverged human populations. Here, we investigate the population genetic history of this region by characterizing patterns of genome-wide variation among 330 individuals from three of the Greater Antilles (Cuba, Puerto Rico, Hispaniola), two mainland (Honduras, Colombia), and three Native South American (Yukpa, Bari, and Warao) populations. We combine these data with a unique database of genomic variation in over 3,000 individuals from diverse European, African, and Native American populations. We use local ancestry inference and tract length distributions to test different demographic scenarios for the pre- and post-colonial history of the region. We develop a novel ancestry-specific PCA (ASPCA) method to reconstruct the sub-continental origin of Native American, European, and African haplotypes from admixed genomes. We find that the most likely source of the indigenous ancestry in Caribbean islanders is a Native South American component shared among inland Amazonian tribes, Central America, and the Yucatan peninsula, suggesting extensive gene flow across the Caribbean in pre-Columbian times. We find evidence of two pulses of African migration. The first pulse—which today is reflected by shorter, older ancestry tracts—consists of a genetic component more similar to coastal West African regions involved in early stages of the trans-Atlantic slave trade. The second pulse—reflected by longer, younger tracts—is more similar to present-day West-Central African populations, supporting historical records of later transatlantic deportation. Surprisingly, we also identify a Latino-specific European component that has significantly diverged from its parental Iberian source populations, presumably as a result of small European founder population size. We demonstrate that the ancestral components in admixed genomes can be traced back to distinct sub-continental source populations with far greater resolution than previously thought, even when limited pre-Columbian Caribbean haplotypes have survived.
Link
European origin of domesticated dogs
It seems like yesterday that a paper suggested a Southeast Asian origin of domestic dogs. It always seems that ancient DNA upsets inferences from modern populations alone.
Science 15 November 2013: Vol. 342 no. 6160 pp. 871-874
Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs
O. Thalmann et al.
The geographic and temporal origins of the domestic dog remain controversial, as genetic data suggest a domestication process in East Asia beginning 15,000 years ago, whereas the oldest doglike fossils are found in Europe and Siberia and date to >30,000 years ago. We analyzed the mitochondrial genomes of 18 prehistoric canids from Eurasia and the New World, along with a comprehensive panel of modern dogs and wolves. The mitochondrial genomes of all modern dogs are phylogenetically most closely related to either ancient or modern canids of Europe. Molecular dating suggests an onset of domestication there 18,800 to 32,100 years ago. These findings imply that domestic dogs are the culmination of a process that initiated with European hunter-gatherers and the canids with whom they interacted.
Link
Science 15 November 2013: Vol. 342 no. 6160 pp. 871-874
Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs
O. Thalmann et al.
The geographic and temporal origins of the domestic dog remain controversial, as genetic data suggest a domestication process in East Asia beginning 15,000 years ago, whereas the oldest doglike fossils are found in Europe and Siberia and date to >30,000 years ago. We analyzed the mitochondrial genomes of 18 prehistoric canids from Eurasia and the New World, along with a comprehensive panel of modern dogs and wolves. The mitochondrial genomes of all modern dogs are phylogenetically most closely related to either ancient or modern canids of Europe. Molecular dating suggests an onset of domestication there 18,800 to 32,100 years ago. These findings imply that domestic dogs are the culmination of a process that initiated with European hunter-gatherers and the canids with whom they interacted.
Link
November 08, 2013
Europeans and South Asians share by descent SLC24A5 light skin allele

Razib has more.
PLoS Genet 9(11): e1003912. doi:10.1371/journal.pgen.1003912
The Light Skin Allele of SLC24A5 in South Asians and Europeans Shares Identity by Descent
Chandana Basu Mallick et al.
Skin pigmentation is one of the most variable phenotypic traits in humans. A non-synonymous substitution (rs1426654) in the third exon of SLC24A5 accounts for lighter skin in Europeans but not in East Asians. A previous genome-wide association study carried out in a heterogeneous sample of UK immigrants of South Asian descent suggested that this gene also contributes significantly to skin pigmentation variation among South Asians. In the present study, we have quantitatively assessed skin pigmentation for a largely homogeneous cohort of 1228 individuals from the Southern region of the Indian subcontinent. Our data confirm significant association of rs1426654 SNP with skin pigmentation, explaining about 27% of total phenotypic variation in the cohort studied. Our extensive survey of the polymorphism in 1573 individuals from 54 ethnic populations across the Indian subcontinent reveals wide presence of the derived-A allele, although the frequencies vary substantially among populations. We also show that the geospatial pattern of this allele is complex, but most importantly, reflects strong influence of language, geography and demographic history of the populations. Sequencing 11.74 kb of SLC24A5 in 95 individuals worldwide reveals that the rs1426654-A alleles in South Asian and West Eurasian populations are monophyletic and occur on the background of a common haplotype that is characterized by low genetic diversity. We date the coalescence of the light skin associated allele at 22–28 KYA. Both our sequence and genome-wide genotype data confirm that this gene has been a target for positive selection among Europeans. However, the latter also shows additional evidence of selection in populations of the Middle East, Central Asia, Pakistan and North India but not in South India.
Link
Early cattle management in NE China

The haplogroup retrieved has so far not been found in modern cattle. However, as mtDNA represents a single genetic locus, it is prone to genetic drift and could easily have been lost by drift even if hybridization between the population to which the Chinese specimen belonged and other domesticated cattle populations has occurred. Further analyses on nuclear DNA will be necessary to show whether this early Chinese cattle management was a short-lived episode or whether it has contributed to the nuclear gene pool of modern cattle.
Nature Communications 4, Article number: 2755 doi:10.1038/ncomms3755
Morphological and genetic evidence for early Holocene cattle management in northeastern China
Hucai Zhang et al.
The domestication of cattle is generally accepted to have taken place in two independent centres: around 10,500 years ago in the Near East, giving rise to modern taurine cattle, and two millennia later in southern Asia, giving rise to zebu cattle. Here we provide firmly dated morphological and genetic evidence for early Holocene management of taurine cattle in northeastern China. We describe conjoining mandibles from this region that show evidence of oral stereotypy, dated to the early Holocene by two independent 14C dates. Using Illumina high-throughput sequencing coupled with DNA hybridization capture, we characterize 15,406 bp of the mitogenome with on average 16.7-fold coverage. Phylogenetic analyses reveal a hitherto unknown mitochondrial haplogroup that falls outside the known taurine diversity. Our data suggest that the first attempts to manage cattle in northern China predate the introduction of domestic cattle that gave rise to the current stock by several thousand years.
Link
November 06, 2013
MEGA6 evolutionary genetics software released
Mol Biol Evol (2013)
doi: 10.1093/molbev/mst197
MEGA6: Molecular Evolutionary Genetics Analysis version 6.0
Koichiro Tamura et al.
We announce the release of an advanced version of the Molecular Evolutionary Genetics Analysis (MEGA) software, which currently contains facilities for building sequence alignments, inferring phylogenetic histories, and conducting molecular evolutionary analysis. In version 6.0, MEGA now enables the inference of timetrees, as it implements the RelTime method for estimating divergence times for all branching points in a phylogeny. A new Timetree Wizard in MEGA6 facilitates this timetree inference by providing a graphical user interface (GUI) to specify the phylogeny and calibration constraints step-by-step. This version also contains enhanced algorithms to search for the optimal trees under evolutionary criteria and implements a more advanced memory management that can double the size of sequence data sets to which MEGA can be applied. Both GUI and command-line versions of MEGA6 can be downloaded from www.megasoftware.net free of charge.
Link
MEGA6: Molecular Evolutionary Genetics Analysis version 6.0
Koichiro Tamura et al.
We announce the release of an advanced version of the Molecular Evolutionary Genetics Analysis (MEGA) software, which currently contains facilities for building sequence alignments, inferring phylogenetic histories, and conducting molecular evolutionary analysis. In version 6.0, MEGA now enables the inference of timetrees, as it implements the RelTime method for estimating divergence times for all branching points in a phylogeny. A new Timetree Wizard in MEGA6 facilitates this timetree inference by providing a graphical user interface (GUI) to specify the phylogeny and calibration constraints step-by-step. This version also contains enhanced algorithms to search for the optimal trees under evolutionary criteria and implements a more advanced memory management that can double the size of sequence data sets to which MEGA can be applied. Both GUI and command-line versions of MEGA6 can be downloaded from www.megasoftware.net free of charge.
Link
Dealing with false positive IBD segments
False positive IBD segments are a real problem for those who wish to use genotype data to establish family connections with distant relatives. Traditionally, this involves finding shared common IBD segments, and then comparing genealogies to find potential common ancestors from which these segments could be inherited. IBD is also used in population genetics (e.g., Coop & Ralph 2013). There is an obvious tradeoff, since sloppy IBD detection may enable more genealogical links to be established but adds to the burden of establishing the validity of these links (the infamous "ignoring contact requests from potential genetic cousins" issue). It will be nice if this technology finds its way to end users who stand to most benefit from it.
arXiv:1311.1120 [q-bio.PE]
Reducing pervasive false positive identical-by-descent segments detected by large-scale pedigree analysis
Eric Y. Durand, Nicholas Eriksson, Cory Y. McLean
(Submitted on 5 Nov 2013)
Analysis of genomic segments shared identical-by-descent (IBD) between individuals is fundamental to many genetic applications, but IBD detection accuracy in non-simulated data is largely unknown. Using 25,432 genotyped European individuals, and exploiting known familial relationships in 2,952 father-mother-child trios contained therein, we identify a false positive rate over 67% for short (2-4 centiMorgan) segments. We introduce a novel, computationally-efficient, haplotype-based metric that enables accurate IBD detection on population-scale datasets.
Link
arXiv:1311.1120 [q-bio.PE]
Reducing pervasive false positive identical-by-descent segments detected by large-scale pedigree analysis
Eric Y. Durand, Nicholas Eriksson, Cory Y. McLean
(Submitted on 5 Nov 2013)
Analysis of genomic segments shared identical-by-descent (IBD) between individuals is fundamental to many genetic applications, but IBD detection accuracy in non-simulated data is largely unknown. Using 25,432 genotyped European individuals, and exploiting known familial relationships in 2,952 father-mother-child trios contained therein, we identify a false positive rate over 67% for short (2-4 centiMorgan) segments. We introduce a novel, computationally-efficient, haplotype-based metric that enables accurate IBD detection on population-scale datasets.
Link
November 05, 2013
Population structure in Thailand

Insight into the Peopling of Mainland Southeast Asia from Thai Population Genetic Structure
Pongsakorn Wangkumhang et al.
There is considerable ethno-linguistic and genetic variation among human populations in Asia, although tracing the origins of this diversity is complicated by migration events. Thailand is at the center of Mainland Southeast Asia (MSEA), a region within Asia that has not been extensively studied. Genetic substructure may exist in the Thai population, since waves of migration from southern China throughout its recent history may have contributed to substantial gene flow. Autosomal SNP data were collated for 438,503 markers from 992 Thai individuals. Using the available self-reported regional origin, four Thai subpopulations genetically distinct from each other and from other Asian populations were resolved by Neighbor-Joining analysis using a 41,569 marker subset. Using an independent Principal Components-based unsupervised clustering approach, four major MSEA subpopulations were resolved in which regional bias was apparent. A major ancestry component was common to these MSEA subpopulations and distinguishes them from other Asian subpopulations. On the other hand, these MSEA subpopulations were admixed with other ancestries, in particular one shared with Chinese. Subpopulation clustering using only Thai individuals and the complete marker set resolved four subpopulations, which are distributed differently across Thailand. A Sino-Thai subpopulation was concentrated in the Central region of Thailand, although this constituted a minority in an otherwise diverse region. Among the most highly differentiated markers which distinguish the Thai subpopulations, several map to regions known to affect phenotypic traits such as skin pigmentation and susceptibility to common diseases. The subpopulation patterns elucidated have important implications for evolutionary and medical genetics. The subpopulation structure within Thailand may reflect the contributions of different migrants throughout the history of MSEA. The information will also be important for genetic association studies to account for population-structure confounding effects.
Link
European pigs replacing Near Eastern ones in Iron Age Israel

Scientific Reports 3, Article number: 3035 doi:10.1038/srep03035
Ancient DNA and Population Turnover in Southern Levantine Pigs- Signature of the Sea Peoples Migration?
Meirav Meiri et al.
Near Eastern wild boars possess a characteristic DNA signature. Unexpectedly, wild boars from Israel have the DNA sequences of European wild boars and domestic pigs. To understand how this anomaly evolved, we sequenced DNA from ancient and modern pigs from Israel. Pigs from Late Bronze Age (until ca. 1150 BCE) in Israel shared haplotypes of modern and ancient Near Eastern pigs. European haplotypes became dominant only during the Iron Age (ca. 900 BCE). This raises the possibility that European pigs were brought to the region by the Sea Peoples who migrated to the Levant at that time. Then, a complete genetic turnover took place, most likely because of repeated admixture between local and introduced European domestic pigs that went feral. Severe population bottlenecks likely accelerated this process. Introductions by humans have strongly affected the phylogeography of wild animals, and interpretations of phylogeography based on modern DNA alone should be taken with caution.
Link
October 30, 2013
Visualizing Y-haplogroup distributions in west Eurasia

The database contains distributions representing 90 populations (N = 16,751 males) by the frequencies of the published and unpublished Y-chromosome Hgs. These Hgs were combined into 18 different Hgs (C, E, ABDF*, G, H, I1, I2, J1, J2, K*, L, N, O, Q, R1a, R1b, R2, T), so that published sources could be used for comparisons.
...
As shown in Fig. 1, Middle Eastern (Class 7) and Central European Classes (Class 8) form one non-separable cluster in the central part of the figure. All of the others of the 10 classes can be identified in different well separable areas around this central region. The Central Asian (Class 4) and Northwest Caucasian (Class 9) Classes are in neighbouring areas in the upper and upper-left parts, while the Arab-Dagestanian Class (Class 1) occupies the opposite, lower-left part of the map. The North-Central and Western European (Class 3 + Class 6) as well as the Atlantic (Class 10) Classes form a common branch in the lower-left part of the figure. The opposite, upper-right branch contains the East Baltic (Class 5) and North Eurasian (Class 2) Classes.Forensic Science International: Genetics Supplement Series Available online 26 October 2013
Classification of the Y-haplogroup distributions of Western Eurasian populations using a self-learning algorithm
H. Pamjav et al.
The understanding of historical relationship between populations is a core aspect of human population history studies. We have compared the frequency of 18 different Y-SNP haplogroups in 90 Western Eurasian populations. Classification of haplogroup distribution vectors using a new self-learning classification algorithm so called “self-organizing cloud (SOC)” proved to be an effective tool to identify population groups, which share common paternal genetic features. By means of the algorithm, we have determined 10 different classes of populations based on the similarity of haplogroup composition. The analysis showed that paternal genetic markers tend to reflect geographical proximity of populations better than linguistic relationship, although certain Y-SNP haplogroups have relatively good correlation with specific language families. These observations are based on the comparative analysis of the Hg distributions of contemporary populations may reflect demographic history of them in the past.
Link
October 29, 2013
Intra-African variation in Neandertal admixture is due to non-African admixture
I haven't read this, but the idea seems to be that variation between Africans in Neandertal admixture can be wholly explained by recent admixture with Eurasians (who already had this type of admixture). This is not very surprising, given that Neandertals were a Eurasian-distributed species, so that admixture with them cannot have taken place in Africa.
The finding that Africans don't vary in their Neandertal admixture suggests that the source cannot have been an unknown African hominin related of Neandertals (in which case we'd expect to see variation in Africans). I don't know of any anthropologically plausible African cousin of the Neandertals, but, of course, the lack of anthropological evidence does not mean non-existence (cf. Denisovans as an anthropologically invisible Neandertal relative in Eurasia).
Genome Biol Evol. 2013 Oct 25. [Epub ahead of print]
Apparent Variation in Neanderthal Admixture among African Populations is Consistent with Gene Flow from non-African Populations.
Wang S, Lachance J, Tishkoff S, Hey J, Xing J.
Abstract
Recent studies have found evidence of introgression from Neanderthals into modern humans outside of sub-Saharan Africa. Given the geographic range of Neanderthals, the findings have been interpreted as evidence of gene exchange between Neanderthals and the modern humans descended from the Out-of-Africa (OOA) migration. Here we examine an alternative interpretation in which the introgression occurred earlier within Africa, between ancestors or relatives of Neanderthals and a subset of African modern humans who were the ancestors of those involved in the OOA migration. Under the alternative model, if the population structure among present-day Africans predates the OOA migration, we might find some African populations show a signal of Neanderthal introgression while others do not. To test this alternative model we compiled a whole-genome data set including 38 sub-Saharan Africans from eight populations and 25 non-African individuals from five populations. We assessed differences in the amount of Neanderthal-like SNP alleles among these populations and observed up to 1.5% difference in the number of Neanderthal-like alleles among African populations. Further analyses suggest that these differences are likely due to recent non-African admixture in these populations. After accounting for recent non-African admixture, our results do not support the alternative model of older (e.g., >100 kya) admixture between modern human and Neanderthal-like hominid within Africa.
Link
The finding that Africans don't vary in their Neandertal admixture suggests that the source cannot have been an unknown African hominin related of Neandertals (in which case we'd expect to see variation in Africans). I don't know of any anthropologically plausible African cousin of the Neandertals, but, of course, the lack of anthropological evidence does not mean non-existence (cf. Denisovans as an anthropologically invisible Neandertal relative in Eurasia).
Genome Biol Evol. 2013 Oct 25. [Epub ahead of print]
Apparent Variation in Neanderthal Admixture among African Populations is Consistent with Gene Flow from non-African Populations.
Wang S, Lachance J, Tishkoff S, Hey J, Xing J.
Abstract
Recent studies have found evidence of introgression from Neanderthals into modern humans outside of sub-Saharan Africa. Given the geographic range of Neanderthals, the findings have been interpreted as evidence of gene exchange between Neanderthals and the modern humans descended from the Out-of-Africa (OOA) migration. Here we examine an alternative interpretation in which the introgression occurred earlier within Africa, between ancestors or relatives of Neanderthals and a subset of African modern humans who were the ancestors of those involved in the OOA migration. Under the alternative model, if the population structure among present-day Africans predates the OOA migration, we might find some African populations show a signal of Neanderthal introgression while others do not. To test this alternative model we compiled a whole-genome data set including 38 sub-Saharan Africans from eight populations and 25 non-African individuals from five populations. We assessed differences in the amount of Neanderthal-like SNP alleles among these populations and observed up to 1.5% difference in the number of Neanderthal-like alleles among African populations. Further analyses suggest that these differences are likely due to recent non-African admixture in these populations. After accounting for recent non-African admixture, our results do not support the alternative model of older (e.g., >100 kya) admixture between modern human and Neanderthal-like hominid within Africa.
Link
Interesting talks @ Penn: Zheng He and Mount Vesuvius
I had recently mentioned Zheng He on account of his Y chromosome.
Great Voyages: Zheng He
Pompeii Lecture Series: Mount Vesuvius in Human History
Great Voyages: Zheng He
Pompeii Lecture Series: Mount Vesuvius in Human History
October 28, 2013
October 26, 2013
Afghan mega-paper (Di Cristofaro et al.)
The admixture results nicely presented on a map:

The authors note that none of the ancestral components peaks in Central Asia, concluding that this region has been a destination rather than a source of population movements. I certainly agree that Central Asia has a lot of recent history affecting it from virtually all directions. On the other hand, we should be cautious about interpreting geographical clines in terms of directionality of population movement; a good example is Sardinia which often emerges as a "focus" of Mediterranean ancestry, but this does not mean that it is the origin of such ancestry. It would certainly be interesting to remove the layers of more recent ancestry from Central Asia to see what was there before the last few thousand years.
The PCA based on autosomal data:

The Y-chromosome haplogroup data can be found in Figure S7. The authors comment:
PLoS ONE 8(10): e76748. doi:10.1371/journal.pone.0076748
Afghan Hindu Kush: Where Eurasian Sub-Continent Gene Flows Converge
Julie Di Cristofaro et al.
Despite being located at the crossroads of Asia, genetics of the Afghanistan populations have been largely overlooked. It is currently inhabited by five major ethnic populations: Pashtun, Tajik, Hazara, Uzbek and Turkmen. Here we present autosomal from a subset of our samples, mitochondrial and Y- chromosome data from over 500 Afghan samples among these 5 ethnic groups. This Afghan data was supplemented with the same Y-chromosome analyses of samples from Iran, Kyrgyzstan, Mongolia and updated Pakistani samples (HGDP-CEPH). The data presented here was integrated into existing knowledge of pan-Eurasian genetic diversity. The pattern of genetic variation, revealed by structure-like and Principal Component analyses and Analysis of Molecular Variance indicates that the people of Afghanistan are made up of a mosaic of components representing various geographic regions of Eurasian ancestry. The absence of a major Central Asian-specific component indicates that the Hindu Kush, like the gene pool of Central Asian populations in general, is a confluence of gene flows rather than a source of distinctly autochthonous populations that have arisen in situ: a conclusion that is reinforced by the phylogeography of both haploid loci.
Link

The authors note that none of the ancestral components peaks in Central Asia, concluding that this region has been a destination rather than a source of population movements. I certainly agree that Central Asia has a lot of recent history affecting it from virtually all directions. On the other hand, we should be cautious about interpreting geographical clines in terms of directionality of population movement; a good example is Sardinia which often emerges as a "focus" of Mediterranean ancestry, but this does not mean that it is the origin of such ancestry. It would certainly be interesting to remove the layers of more recent ancestry from Central Asia to see what was there before the last few thousand years.
The PCA based on autosomal data:

The Y-chromosome haplogroup data can be found in Figure S7. The authors comment:
94% of the chromosomes are distributed within the following 9 main haplogroups: R-M207 (34%), J-M304 (16%), C-M130 (15%), L-M20 (6%), G-M201 (6%), Q-M242 (6%), N-M231 (4%), O-M175 (4%) and E-M96 (3%). Within the core haplogroups observed in the Afghan populations, there are sub-haplogroups that provide more refined insights into the underlying structure of the Y-chromosome gene pool. One of the important sub-haplogroups includes the C3b2b1-M401 lineage that is amplified in Hazara, Kyrgyz and Mongol populations. Haplogroup G2c-M377 reaches 14.7% in Pashtun, consistent with previous results [31], whereas it is virtually absent from all other populations. J2a1-Page55 is found in 23% of Iranians, 13% of the Hazara from the Hindu Kush, 11% of the Tajik and Uzbek from the Hindu Kush, 10% of Pakistanis, 4% of the Turkmen from the Hindu Kush, 3% of the Pashtun and 2% of the Kyrgyz and Mongol populations. Concerning haplogroup L, L1c-M357 is significantly higher in Burusho and Kalash (15% and 25%) than in other populations. L1a-M76 is most frequent in Balochi (20%), and is found at lower levels in Kyrgyz, Pashtun, Tajik, Uzbek and Turkmen populations. Q1a2-M25 lineage is characteristic of Turkmen (31%), significantly higher than all other populations. Haplogroup R1a1a-M198/M17 is characterized by its absence or very low frequency in Iranian, Mongol and Hazara populations and its high frequency in Pashtun and Kyrgyz populations.
PLoS ONE 8(10): e76748. doi:10.1371/journal.pone.0076748
Afghan Hindu Kush: Where Eurasian Sub-Continent Gene Flows Converge
Julie Di Cristofaro et al.
Despite being located at the crossroads of Asia, genetics of the Afghanistan populations have been largely overlooked. It is currently inhabited by five major ethnic populations: Pashtun, Tajik, Hazara, Uzbek and Turkmen. Here we present autosomal from a subset of our samples, mitochondrial and Y- chromosome data from over 500 Afghan samples among these 5 ethnic groups. This Afghan data was supplemented with the same Y-chromosome analyses of samples from Iran, Kyrgyzstan, Mongolia and updated Pakistani samples (HGDP-CEPH). The data presented here was integrated into existing knowledge of pan-Eurasian genetic diversity. The pattern of genetic variation, revealed by structure-like and Principal Component analyses and Analysis of Molecular Variance indicates that the people of Afghanistan are made up of a mosaic of components representing various geographic regions of Eurasian ancestry. The absence of a major Central Asian-specific component indicates that the Hindu Kush, like the gene pool of Central Asian populations in general, is a confluence of gene flows rather than a source of distinctly autochthonous populations that have arisen in situ: a conclusion that is reinforced by the phylogeography of both haploid loci.
Link
New aDNA capture method (plus some data on ancient individuals from Bulgaria, Denmark, and Peru)
This seems to present an alternative method for capture of ancient DNA libraries than the one used on the Tianyuan individual. It is mostly a methods paper, but also has some initial analysis of some ancient individuals. From the paper:
Also:
Hopefully a more complete analysis of this data and production of more data with this method will follow in the future.
The American Journal of Human Genetics (2013), http://dx.doi.org/10.1016/j.ajhg.2013.10.002
Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries
Meredith L. Carpenter et al.
Most ancient specimens contain very low levels of endogenous DNA, precluding the shotgun sequencing of many interesting samples because of cost. Ancient DNA (aDNA) libraries often contain less than 1% endogenous DNA, with the majority of sequencing capacity taken up by environmental DNA. Here we present a capture-based method for enriching the endogenous component of aDNA sequencing libraries. By using biotinylated RNA baits transcribed from genomic DNA libraries, we are able to capture DNA fragments from across the human genome. We demonstrate this method on libraries created from four Iron Age and Bronze Age human teeth from Bulgaria, as well as bone samples from seven Peruvian mummies and a Bronze Age hair sample from Denmark. Prior to capture, shotgun sequencing of these libraries yielded an average of 1.2% of reads mapping to the human genome (including duplicates). After capture, this fraction increased substantially, with up to 59% of reads mapped to human and enrichment ranging from 6- to 159-fold. Furthermore, we maintained coverage of the majority of regions sequenced in the precapture library. Intersection with the 1000 Genomes Project reference panel yielded an average of 50,723 SNPs (range 3,062–147,243) for the postcapture libraries sequenced with 1 million reads, compared with 13,280 SNPs (range 217–73,266) for the precapture libraries, increasing resolution in population genetic analyses. Our whole-genome capture approach makes it less costly to sequence aDNA from specimens containing very low levels of endogenous DNA, enabling the analysis of larger numbers of samples.
Link (pdf)
We were able to tentatively call mtDNA haplogroups for these samples (Table S1). The two Bulgarian Iron Age individuals (P192-1 and T2G5) fell into haplogroups U3b and HV(16311), respectively. Haplogroup U3 is especially common in the countries surrounding the Black Sea, including Bulgaria, and in the Near East, and HV is also found at low frequencies in Europe and peaks in the Near East.41 The three Peruvian mummies fell into haplogroups B2, M (an ancestor of D), and D1, all derived from founder Native American lineages and previously observed in both pre-Columbian and modern populations from Peru.
Also:
For the Peruvian mummies, we also included 10 Native American individuals from Central and South America in the PCA (Figures 3E and 3F). Interestingly, all of the mummies fell between the Native American populations (KAR, MAY, AYM) and East Asian populations (JPT, CHS, CHB), as would be expected for a nonadmixed Native American individual (Figures 3E, 3F, and S2). These mummies belonged to the pre-Columbian Chachapoya culture, who, by some accounts, were unusually fair-skinned,39 suggesting a potential for pre- Columbian European admixture. However, based on our preliminary results, these individuals appear to have been ancestrally Native American.The Peruvian mummies were from 1000-1500AD, so it's not very surprising that they don't appear to have European admixture and to be "ancestrally Native American".
Hopefully a more complete analysis of this data and production of more data with this method will follow in the future.
The American Journal of Human Genetics (2013), http://dx.doi.org/10.1016/j.ajhg.2013.10.002
Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries
Meredith L. Carpenter et al.
Most ancient specimens contain very low levels of endogenous DNA, precluding the shotgun sequencing of many interesting samples because of cost. Ancient DNA (aDNA) libraries often contain less than 1% endogenous DNA, with the majority of sequencing capacity taken up by environmental DNA. Here we present a capture-based method for enriching the endogenous component of aDNA sequencing libraries. By using biotinylated RNA baits transcribed from genomic DNA libraries, we are able to capture DNA fragments from across the human genome. We demonstrate this method on libraries created from four Iron Age and Bronze Age human teeth from Bulgaria, as well as bone samples from seven Peruvian mummies and a Bronze Age hair sample from Denmark. Prior to capture, shotgun sequencing of these libraries yielded an average of 1.2% of reads mapping to the human genome (including duplicates). After capture, this fraction increased substantially, with up to 59% of reads mapped to human and enrichment ranging from 6- to 159-fold. Furthermore, we maintained coverage of the majority of regions sequenced in the precapture library. Intersection with the 1000 Genomes Project reference panel yielded an average of 50,723 SNPs (range 3,062–147,243) for the postcapture libraries sequenced with 1 million reads, compared with 13,280 SNPs (range 217–73,266) for the precapture libraries, increasing resolution in population genetic analyses. Our whole-genome capture approach makes it less costly to sequence aDNA from specimens containing very low levels of endogenous DNA, enabling the analysis of larger numbers of samples.
Link (pdf)