A currently not available preprint that has important implications about the Neolithic of Europe.
A late Neolithic Iberian farmer exhibits genetic affinity to Neolithic Scandinavian farmers and a Bronze Age central European farmer
Sverrisdóttir, Oddný Ósk et al.
The spread of farming, the neolithisation process, swept over Europe after the advent of the farming lifestyle in the near east approximately 11,000 years ago. However the mode of transmission and its impact on the demographic patterns of Europe remains largely unknown. In this study we obtained : 66,476,944 bp of genomic DNA from the remains of a 4000 year old Neolithic farmer from the site of El Portalón, 15 km east of Burgos, Spain. We compared the genomic signature of this individual to modern-day populations as well as the few Neolithic individuals that has produced large-scale autosomal data. The Neolithic Portalón individual is genetically most similar to southern Europeans, similar to a Scandinavian Neolithic farmer and the Tyrolean Iceman. In contrast, the Neolithic Portalón individual displays little affinity to two Mesolithic samples from the near-by area, La Brana, demonstrating a distinct change in population history between 7,000 and 4,000 years ago for the northern Iberian Peninsula.
Link

November 28, 2013
November 26, 2013
One to three men fathered most western Europeans?
It may sound far-fetched but it's certainly possible. After all, no R1b has been found in Europe before a Bell Beaker site from the 3rd millennium BC and today many Europeans (most in western Europe) belong to this haplogroup. As more Y chromosomes are sampled from ancient Europe, it will become clear if the R1b frequency actually shot from non-existence to ubiquity over a short span of time, and the Y chromosomes after the transition will be practically clones of each other.
Investigative Genetics 2013, 4:25 doi:10.1186/2041-2223-4-25
Modeling the contrasting Neolithic male lineage expansions in Europe and Africa
Michael J Sikora et al.
Abstract (provisional)
Background
Patterns of genetic variation in a population carry information about the prehistory of the population, and for the human Y chromosome an especially informative phylogenetic tree has previously been constructed from fully-sequenced chromosomes. This revealed contrasting bifurcating and starlike phylogenies for the major lineages associated with the Neolithic expansions in sub-Saharan Africa and Western Europe, respectively.
Results
We used coalescent simulations to investigate the range of demographic models most likely to produce the phylogenetic structures observed in Africa and Europe, assessing the starting and ending genetic effective population sizes, duration of the expansion, and time when expansion ended. The best-fitting models in Africa and Europe are very different. In Africa, the expansion took about 12 thousand years, ending very recently; it started from approximately 40 men and numbers expanded approximately 50-fold. In Europe, the expansion was much more rapid, taking only a few generations and occurring as soon as the major R1b lineage entered Europe; it started from just one to three men, whose numbers expanded more than a thousandfold.
Conclusions
Although highly simplified, the demographic model we have used captures key elements of the differences between the male Neolithic expansions in Africa and Europe, and is consistent with archaeological findings.
Link
Investigative Genetics 2013, 4:25 doi:10.1186/2041-2223-4-25
Modeling the contrasting Neolithic male lineage expansions in Europe and Africa
Michael J Sikora et al.
Abstract (provisional)
Background
Patterns of genetic variation in a population carry information about the prehistory of the population, and for the human Y chromosome an especially informative phylogenetic tree has previously been constructed from fully-sequenced chromosomes. This revealed contrasting bifurcating and starlike phylogenies for the major lineages associated with the Neolithic expansions in sub-Saharan Africa and Western Europe, respectively.
Results
We used coalescent simulations to investigate the range of demographic models most likely to produce the phylogenetic structures observed in Africa and Europe, assessing the starting and ending genetic effective population sizes, duration of the expansion, and time when expansion ended. The best-fitting models in Africa and Europe are very different. In Africa, the expansion took about 12 thousand years, ending very recently; it started from approximately 40 men and numbers expanded approximately 50-fold. In Europe, the expansion was much more rapid, taking only a few generations and occurring as soon as the major R1b lineage entered Europe; it started from just one to three men, whose numbers expanded more than a thousandfold.
Conclusions
Although highly simplified, the demographic model we have used captures key elements of the differences between the male Neolithic expansions in Africa and Europe, and is consistent with archaeological findings.
Link
A priori Y chromosome phylogeny from sequencing data
A cool new paper by a team of citizen scientists. The most important new piece of evidence is the joining together of haplogroup M (Papuans) with P in a new MP internal node. Your guess is as good as mine as to whether this MP may have come from, as his descendants are presently spread from the Atlantic via Siberia to the Amazon and all the way to New Guinea. The Mal'ta boy belonged to haplogroup R.
The other interesting discovery is of one Telugu man from India who shares mutations with haplogroups N and O but belongs to neither N nor O, so this defines a new "X" clade in the phylogeny. I am wondering if this could perhaps be called NO0 instead, similar to the way that more basal clades of the entire phylogeny were called A0, A00, and so on? Terminology is tricky...

I am aware of a few commercial ventures to resequence Y chromosomes, and I'm pretty sure that citizen scientists will soon not only be able to re-analyze data such as those from the 1000 Genomes Project, but will be able to generate data of their own.
bioRxiv doi: 10.1101/000802
Generation of high-resolution a priori Y-chromosome phylogenies using “next-generation” sequencing data
Gregory R Magoon et al.
An approach for generating high-resolution a priori maximum parsimony Y-chromosome (“chrY”) phylogenies based on SNP and small INDEL variant data from massively-parallel short-read (“next-generation”) sequencing data is described; the tree-generation methodology produces annotations localizing mutations to individual branches of the tree, along with indications of mutation placement uncertainty in cases for which "no-calls" (through lack of mapped reads or otherwise) at particular site precludes a precise placement of the mutation. The approach leverages careful variant site filtering and a novel iterative reweighting procedure to generate high-accuracy trees while considering variants in regions of chrY that had previously been excluded from analyses based on short-read sequencing data. It is argued that the proposed approach is also superior to previous region-based filtering approaches in that it adapts to the quality of the underlying data and will automatically allow the scope of sites considered to expand as the underlying data quality (e.g. through longer read lengths) improves. Key related issues, including calling of genotypes for the hemizygous chrY, reliability of variant results, read mismappings and "heterozygous" genotype calls, and the mutational stability of different variants are discussed and taken into account. The methodology is demonstrated through application to a dataset consisting of 1292 male samples from diverse populations and haplogroups, with the majority coming from low-coverage sequencing by the 1000 Genomes Project. Application of the tree-generation approach to these data produces a tree involving over 120,000 chrY variant sites (about 45,000 sites if “singletons” are excluded). The utility of this approach in refining the Y-chromosome phylogenetic tree is demonstrated by examining results for several haplogroups. The results indicate a number of new branches on the Y-chromosome phylogenetic tree, many of them subdividing known branches, but also including some that inform the presence of additional levels along the “trunk” of the tree. Finally, opportunities for extensions of this phylogenetic analysis approach to other types of genetic data are examined.
Link
The other interesting discovery is of one Telugu man from India who shares mutations with haplogroups N and O but belongs to neither N nor O, so this defines a new "X" clade in the phylogeny. I am wondering if this could perhaps be called NO0 instead, similar to the way that more basal clades of the entire phylogeny were called A0, A00, and so on? Terminology is tricky...

I am aware of a few commercial ventures to resequence Y chromosomes, and I'm pretty sure that citizen scientists will soon not only be able to re-analyze data such as those from the 1000 Genomes Project, but will be able to generate data of their own.
bioRxiv doi: 10.1101/000802
Generation of high-resolution a priori Y-chromosome phylogenies using “next-generation” sequencing data
Gregory R Magoon et al.
An approach for generating high-resolution a priori maximum parsimony Y-chromosome (“chrY”) phylogenies based on SNP and small INDEL variant data from massively-parallel short-read (“next-generation”) sequencing data is described; the tree-generation methodology produces annotations localizing mutations to individual branches of the tree, along with indications of mutation placement uncertainty in cases for which "no-calls" (through lack of mapped reads or otherwise) at particular site precludes a precise placement of the mutation. The approach leverages careful variant site filtering and a novel iterative reweighting procedure to generate high-accuracy trees while considering variants in regions of chrY that had previously been excluded from analyses based on short-read sequencing data. It is argued that the proposed approach is also superior to previous region-based filtering approaches in that it adapts to the quality of the underlying data and will automatically allow the scope of sites considered to expand as the underlying data quality (e.g. through longer read lengths) improves. Key related issues, including calling of genotypes for the hemizygous chrY, reliability of variant results, read mismappings and "heterozygous" genotype calls, and the mutational stability of different variants are discussed and taken into account. The methodology is demonstrated through application to a dataset consisting of 1292 male samples from diverse populations and haplogroups, with the majority coming from low-coverage sequencing by the 1000 Genomes Project. Application of the tree-generation approach to these data produces a tree involving over 120,000 chrY variant sites (about 45,000 sites if “singletons” are excluded). The utility of this approach in refining the Y-chromosome phylogenetic tree is demonstrated by examining results for several haplogroups. The results indicate a number of new branches on the Y-chromosome phylogenetic tree, many of them subdividing known branches, but also including some that inform the presence of additional levels along the “trunk” of the tree. Finally, opportunities for extensions of this phylogenetic analysis approach to other types of genetic data are examined.
Link
November 20, 2013
Ancient DNA from Upper Paleolithic Lake Baikal (Mal'ta and Afontova Gora)
The study I mentioned in a previous post has now been made available in Nature. Two Upper Paleolithic Siberians (24-17kya) have been sequenced at low coverage. The better quality (and older) Mal'ta (MA-1) sample belongs to Y-haplogroup R and mtDNA haplogroup U, and the younger (but poorer quality) Afontova Gora (AG-2) sample appears to be related to it.
 Most interestingly, there is evidence for gene flow between the MA-1 sample and Native Americans, which makes sense as these are Siberians of the period leading up to the initial colonization of the Americas. The interesting thing is that MA-1 does not appear to be East Eurasian, as proven by the test D(Papuan, Han; Sardinian, MA-1) which is non-significant, so MA-1 is not more closely related to Han than to Papuans (which is true for modern native Americans). So, it seems that the gene flow between MA-1 and Native Americans was towards Native Americans from MA-1 and not vice versa.
Most interestingly, there is evidence for gene flow between the MA-1 sample and Native Americans, which makes sense as these are Siberians of the period leading up to the initial colonization of the Americas. The interesting thing is that MA-1 does not appear to be East Eurasian, as proven by the test D(Papuan, Han; Sardinian, MA-1) which is non-significant, so MA-1 is not more closely related to Han than to Papuans (which is true for modern native Americans). So, it seems that the gene flow between MA-1 and Native Americans was towards Native Americans from MA-1 and not vice versa.
It is fascinating that such a sample could be found so far east at so early a time. Both Y-chromosome R and mtDNA haplogroup U are very rare east of Lake Baikal which has been considered a limit of west Eurasian influence into east Eurasia. And, indeed, both these haplogroups are absent in Native Americans, so it is not yet clear how Native Americans (who belong to Y-chromosome haplogroups Q and C and mtDNA haplogroups A, B, C, D, X) are related to these Paleolithic Siberians. The obvious candidate for this relationship is Y-chromosome haplogroup P (the ancestor of Q and R). So, perhaps Q-bearing relatives of the R-bearing Mal'ta population settled the Americas.
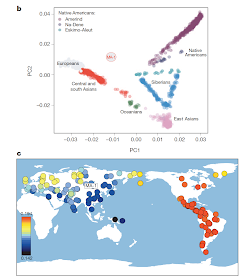 In any case, this is an extremely important sample, as its position in "no man's land" in the PCA plot (left) demonstrates, between Europeans and native Americans but close to no modern population.
In any case, this is an extremely important sample, as its position in "no man's land" in the PCA plot (left) demonstrates, between Europeans and native Americans but close to no modern population.
Its closest present-day relatives are indicated in (c), with Native Americans (red) being the closest, and a scattering of boreal populations from the Atlantic to the Pacific (but not in the vicinity of Lake Baikal) next in line (yellow).
This distribution clearly related to the evidence for admixture in Europe adduced in two other recent papers, although the question of who went where and when remains to be resolved. Was MA-1 part of an intrusive western population encroaching on east Eurasians? Or did MA-1 lookalikes arrive as first settlers in empty territory, later ceding this space to east Eurasians from, perhaps, China? Did the two mix in Siberia or did they arrive in the Americas in separate migrations and mix there? And, how does this all relate to events in Europe in the far west?
UPDATE: Razib makes an excellent point:
Nature (2013) doi:10.1038/nature12736
Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans
Maanasa Raghavan, Pontus Skoglund et al.
The origins of the First Americans remain contentious. Although Native Americans seem to be genetically most closely related to east Asians1, 2, 3, there is no consensus with regard to which specific Old World populations they are closest to4, 5, 6, 7, 8. Here we sequence the draft genome of an approximately 24,000-year-old individual (MA-1), from Mal’ta in south-central Siberia9, to an average depth of 1×. To our knowledge this is the oldest anatomically modern human genome reported to date. The MA-1 mitochondrial genome belongs to haplogroup U, which has also been found at high frequency among Upper Palaeolithic and Mesolithic European hunter-gatherers10, 11, 12, and the Y chromosome of MA-1 is basal to modern-day western Eurasians and near the root of most Native American lineages5. Similarly, we find autosomal evidence that MA-1 is basal to modern-day western Eurasians and genetically closely related to modern-day Native Americans, with no close affinity to east Asians. This suggests that populations related to contemporary western Eurasians had a more north-easterly distribution 24,000 years ago than commonly thought. Furthermore, we estimate that 14 to 38% of Native American ancestry may originate through gene flow from this ancient population. This is likely to have occurred after the divergence of Native American ancestors from east Asian ancestors, but before the diversification of Native American populations in the New World. Gene flow from the MA-1 lineage into Native American ancestors could explain why several crania from the First Americans have been reported as bearing morphological characteristics that do not resemble those of east Asians2, 13. Sequencing of another south-central Siberian, Afontova Gora-2 dating to approximately 17,000 years ago14, revealed similar autosomal genetic signatures as MA-1, suggesting that the region was continuously occupied by humans throughout the Last Glacial Maximum. Our findings reveal that western Eurasian genetic signatures in modern-day Native Americans derive not only from post-Columbian admixture, as commonly thought, but also from a mixed ancestry of the First Americans.
Link

It is fascinating that such a sample could be found so far east at so early a time. Both Y-chromosome R and mtDNA haplogroup U are very rare east of Lake Baikal which has been considered a limit of west Eurasian influence into east Eurasia. And, indeed, both these haplogroups are absent in Native Americans, so it is not yet clear how Native Americans (who belong to Y-chromosome haplogroups Q and C and mtDNA haplogroups A, B, C, D, X) are related to these Paleolithic Siberians. The obvious candidate for this relationship is Y-chromosome haplogroup P (the ancestor of Q and R). So, perhaps Q-bearing relatives of the R-bearing Mal'ta population settled the Americas.

Its closest present-day relatives are indicated in (c), with Native Americans (red) being the closest, and a scattering of boreal populations from the Atlantic to the Pacific (but not in the vicinity of Lake Baikal) next in line (yellow).
This distribution clearly related to the evidence for admixture in Europe adduced in two other recent papers, although the question of who went where and when remains to be resolved. Was MA-1 part of an intrusive western population encroaching on east Eurasians? Or did MA-1 lookalikes arrive as first settlers in empty territory, later ceding this space to east Eurasians from, perhaps, China? Did the two mix in Siberia or did they arrive in the Americas in separate migrations and mix there? And, how does this all relate to events in Europe in the far west?
UPDATE: Razib makes an excellent point:
Also, can we now finally bury the debate when east and west Eurasians diverged? Obviously it can’t have been that recent if a >20,000 year old individual had closer affinity to western populations.We already knew that Tianyuan was more Asian than European, so I think west Eurasians diverged from the rest >40 thousand years ago. But, Tianyuan was so early that its precise relationships to different Asian groups could not be determined. So, I'd say it's a good guess that east-west split off before 40 thousand years in Eurasia.
Nature (2013) doi:10.1038/nature12736
Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans
Maanasa Raghavan, Pontus Skoglund et al.
The origins of the First Americans remain contentious. Although Native Americans seem to be genetically most closely related to east Asians1, 2, 3, there is no consensus with regard to which specific Old World populations they are closest to4, 5, 6, 7, 8. Here we sequence the draft genome of an approximately 24,000-year-old individual (MA-1), from Mal’ta in south-central Siberia9, to an average depth of 1×. To our knowledge this is the oldest anatomically modern human genome reported to date. The MA-1 mitochondrial genome belongs to haplogroup U, which has also been found at high frequency among Upper Palaeolithic and Mesolithic European hunter-gatherers10, 11, 12, and the Y chromosome of MA-1 is basal to modern-day western Eurasians and near the root of most Native American lineages5. Similarly, we find autosomal evidence that MA-1 is basal to modern-day western Eurasians and genetically closely related to modern-day Native Americans, with no close affinity to east Asians. This suggests that populations related to contemporary western Eurasians had a more north-easterly distribution 24,000 years ago than commonly thought. Furthermore, we estimate that 14 to 38% of Native American ancestry may originate through gene flow from this ancient population. This is likely to have occurred after the divergence of Native American ancestors from east Asian ancestors, but before the diversification of Native American populations in the New World. Gene flow from the MA-1 lineage into Native American ancestors could explain why several crania from the First Americans have been reported as bearing morphological characteristics that do not resemble those of east Asians2, 13. Sequencing of another south-central Siberian, Afontova Gora-2 dating to approximately 17,000 years ago14, revealed similar autosomal genetic signatures as MA-1, suggesting that the region was continuously occupied by humans throughout the Last Glacial Maximum. Our findings reveal that western Eurasian genetic signatures in modern-day Native Americans derive not only from post-Columbian admixture, as commonly thought, but also from a mixed ancestry of the First Americans.
Link
November 18, 2013
Royal Society Ancient DNA meeting
Ancient DNA: the first three decades
Ancient DNA: applications in human evolutionary history (this also has abstracts)
Program for both days (pdf). I've seen some tweets from it using the #ancientDNA tag.
Ancient DNA: applications in human evolutionary history (this also has abstracts)
Program for both days (pdf). I've seen some tweets from it using the #ancientDNA tag.
November 15, 2013
Music and population structure (Brown et al. 2013)
Proceedings of the Royal Society B doi: 10.1098/rspb.2013.2072
Correlations in the population structure of music, genes and language
Steven Brown et al.
We present, to our knowledge, the first quantitative evidence that music and genes may have coevolved by demonstrating significant correlations between traditional group-level folk songs and mitochondrial DNA variation among nine indigenous populations of Taiwan. These correlations were of comparable magnitude to those between language and genes for the same populations, although music and language were not significantly correlated with one another. An examination of population structure for genetics showed stronger parallels to music than to language. Overall, the results suggest that music might have a sufficient time-depth to retrace ancient population movements and, additionally, that it might be capturing different aspects of population history than language. Music may therefore have the potential to serve as a novel marker of human migrations to complement genes, language and other markers.
Link
Correlations in the population structure of music, genes and language
Steven Brown et al.
We present, to our knowledge, the first quantitative evidence that music and genes may have coevolved by demonstrating significant correlations between traditional group-level folk songs and mitochondrial DNA variation among nine indigenous populations of Taiwan. These correlations were of comparable magnitude to those between language and genes for the same populations, although music and language were not significantly correlated with one another. An examination of population structure for genetics showed stronger parallels to music than to language. Overall, the results suggest that music might have a sufficient time-depth to retrace ancient population movements and, additionally, that it might be capturing different aspects of population history than language. Music may therefore have the potential to serve as a novel marker of human migrations to complement genes, language and other markers.
Link
Population history of the Caribbean

Reconstructing the Population Genetic History of the Caribbean
Andrés Moreno-Estrada et al.
The Caribbean basin is home to some of the most complex interactions in recent history among previously diverged human populations. Here, we investigate the population genetic history of this region by characterizing patterns of genome-wide variation among 330 individuals from three of the Greater Antilles (Cuba, Puerto Rico, Hispaniola), two mainland (Honduras, Colombia), and three Native South American (Yukpa, Bari, and Warao) populations. We combine these data with a unique database of genomic variation in over 3,000 individuals from diverse European, African, and Native American populations. We use local ancestry inference and tract length distributions to test different demographic scenarios for the pre- and post-colonial history of the region. We develop a novel ancestry-specific PCA (ASPCA) method to reconstruct the sub-continental origin of Native American, European, and African haplotypes from admixed genomes. We find that the most likely source of the indigenous ancestry in Caribbean islanders is a Native South American component shared among inland Amazonian tribes, Central America, and the Yucatan peninsula, suggesting extensive gene flow across the Caribbean in pre-Columbian times. We find evidence of two pulses of African migration. The first pulse—which today is reflected by shorter, older ancestry tracts—consists of a genetic component more similar to coastal West African regions involved in early stages of the trans-Atlantic slave trade. The second pulse—reflected by longer, younger tracts—is more similar to present-day West-Central African populations, supporting historical records of later transatlantic deportation. Surprisingly, we also identify a Latino-specific European component that has significantly diverged from its parental Iberian source populations, presumably as a result of small European founder population size. We demonstrate that the ancestral components in admixed genomes can be traced back to distinct sub-continental source populations with far greater resolution than previously thought, even when limited pre-Columbian Caribbean haplotypes have survived.
Link
European origin of domesticated dogs
It seems like yesterday that a paper suggested a Southeast Asian origin of domestic dogs. It always seems that ancient DNA upsets inferences from modern populations alone.
Science 15 November 2013: Vol. 342 no. 6160 pp. 871-874
Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs
O. Thalmann et al.
The geographic and temporal origins of the domestic dog remain controversial, as genetic data suggest a domestication process in East Asia beginning 15,000 years ago, whereas the oldest doglike fossils are found in Europe and Siberia and date to >30,000 years ago. We analyzed the mitochondrial genomes of 18 prehistoric canids from Eurasia and the New World, along with a comprehensive panel of modern dogs and wolves. The mitochondrial genomes of all modern dogs are phylogenetically most closely related to either ancient or modern canids of Europe. Molecular dating suggests an onset of domestication there 18,800 to 32,100 years ago. These findings imply that domestic dogs are the culmination of a process that initiated with European hunter-gatherers and the canids with whom they interacted.
Link
Science 15 November 2013: Vol. 342 no. 6160 pp. 871-874
Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs
O. Thalmann et al.
The geographic and temporal origins of the domestic dog remain controversial, as genetic data suggest a domestication process in East Asia beginning 15,000 years ago, whereas the oldest doglike fossils are found in Europe and Siberia and date to >30,000 years ago. We analyzed the mitochondrial genomes of 18 prehistoric canids from Eurasia and the New World, along with a comprehensive panel of modern dogs and wolves. The mitochondrial genomes of all modern dogs are phylogenetically most closely related to either ancient or modern canids of Europe. Molecular dating suggests an onset of domestication there 18,800 to 32,100 years ago. These findings imply that domestic dogs are the culmination of a process that initiated with European hunter-gatherers and the canids with whom they interacted.
Link
November 08, 2013
Europeans and South Asians share by descent SLC24A5 light skin allele

Razib has more.
PLoS Genet 9(11): e1003912. doi:10.1371/journal.pgen.1003912
The Light Skin Allele of SLC24A5 in South Asians and Europeans Shares Identity by Descent
Chandana Basu Mallick et al.
Skin pigmentation is one of the most variable phenotypic traits in humans. A non-synonymous substitution (rs1426654) in the third exon of SLC24A5 accounts for lighter skin in Europeans but not in East Asians. A previous genome-wide association study carried out in a heterogeneous sample of UK immigrants of South Asian descent suggested that this gene also contributes significantly to skin pigmentation variation among South Asians. In the present study, we have quantitatively assessed skin pigmentation for a largely homogeneous cohort of 1228 individuals from the Southern region of the Indian subcontinent. Our data confirm significant association of rs1426654 SNP with skin pigmentation, explaining about 27% of total phenotypic variation in the cohort studied. Our extensive survey of the polymorphism in 1573 individuals from 54 ethnic populations across the Indian subcontinent reveals wide presence of the derived-A allele, although the frequencies vary substantially among populations. We also show that the geospatial pattern of this allele is complex, but most importantly, reflects strong influence of language, geography and demographic history of the populations. Sequencing 11.74 kb of SLC24A5 in 95 individuals worldwide reveals that the rs1426654-A alleles in South Asian and West Eurasian populations are monophyletic and occur on the background of a common haplotype that is characterized by low genetic diversity. We date the coalescence of the light skin associated allele at 22–28 KYA. Both our sequence and genome-wide genotype data confirm that this gene has been a target for positive selection among Europeans. However, the latter also shows additional evidence of selection in populations of the Middle East, Central Asia, Pakistan and North India but not in South India.
Link
Early cattle management in NE China

The haplogroup retrieved has so far not been found in modern cattle. However, as mtDNA represents a single genetic locus, it is prone to genetic drift and could easily have been lost by drift even if hybridization between the population to which the Chinese specimen belonged and other domesticated cattle populations has occurred. Further analyses on nuclear DNA will be necessary to show whether this early Chinese cattle management was a short-lived episode or whether it has contributed to the nuclear gene pool of modern cattle.
Nature Communications 4, Article number: 2755 doi:10.1038/ncomms3755
Morphological and genetic evidence for early Holocene cattle management in northeastern China
Hucai Zhang et al.
The domestication of cattle is generally accepted to have taken place in two independent centres: around 10,500 years ago in the Near East, giving rise to modern taurine cattle, and two millennia later in southern Asia, giving rise to zebu cattle. Here we provide firmly dated morphological and genetic evidence for early Holocene management of taurine cattle in northeastern China. We describe conjoining mandibles from this region that show evidence of oral stereotypy, dated to the early Holocene by two independent 14C dates. Using Illumina high-throughput sequencing coupled with DNA hybridization capture, we characterize 15,406 bp of the mitogenome with on average 16.7-fold coverage. Phylogenetic analyses reveal a hitherto unknown mitochondrial haplogroup that falls outside the known taurine diversity. Our data suggest that the first attempts to manage cattle in northern China predate the introduction of domestic cattle that gave rise to the current stock by several thousand years.
Link
November 06, 2013
MEGA6 evolutionary genetics software released
Mol Biol Evol (2013)
doi: 10.1093/molbev/mst197
MEGA6: Molecular Evolutionary Genetics Analysis version 6.0
Koichiro Tamura et al.
We announce the release of an advanced version of the Molecular Evolutionary Genetics Analysis (MEGA) software, which currently contains facilities for building sequence alignments, inferring phylogenetic histories, and conducting molecular evolutionary analysis. In version 6.0, MEGA now enables the inference of timetrees, as it implements the RelTime method for estimating divergence times for all branching points in a phylogeny. A new Timetree Wizard in MEGA6 facilitates this timetree inference by providing a graphical user interface (GUI) to specify the phylogeny and calibration constraints step-by-step. This version also contains enhanced algorithms to search for the optimal trees under evolutionary criteria and implements a more advanced memory management that can double the size of sequence data sets to which MEGA can be applied. Both GUI and command-line versions of MEGA6 can be downloaded from www.megasoftware.net free of charge.
Link
MEGA6: Molecular Evolutionary Genetics Analysis version 6.0
Koichiro Tamura et al.
We announce the release of an advanced version of the Molecular Evolutionary Genetics Analysis (MEGA) software, which currently contains facilities for building sequence alignments, inferring phylogenetic histories, and conducting molecular evolutionary analysis. In version 6.0, MEGA now enables the inference of timetrees, as it implements the RelTime method for estimating divergence times for all branching points in a phylogeny. A new Timetree Wizard in MEGA6 facilitates this timetree inference by providing a graphical user interface (GUI) to specify the phylogeny and calibration constraints step-by-step. This version also contains enhanced algorithms to search for the optimal trees under evolutionary criteria and implements a more advanced memory management that can double the size of sequence data sets to which MEGA can be applied. Both GUI and command-line versions of MEGA6 can be downloaded from www.megasoftware.net free of charge.
Link
Dealing with false positive IBD segments
False positive IBD segments are a real problem for those who wish to use genotype data to establish family connections with distant relatives. Traditionally, this involves finding shared common IBD segments, and then comparing genealogies to find potential common ancestors from which these segments could be inherited. IBD is also used in population genetics (e.g., Coop & Ralph 2013). There is an obvious tradeoff, since sloppy IBD detection may enable more genealogical links to be established but adds to the burden of establishing the validity of these links (the infamous "ignoring contact requests from potential genetic cousins" issue). It will be nice if this technology finds its way to end users who stand to most benefit from it.
arXiv:1311.1120 [q-bio.PE]
Reducing pervasive false positive identical-by-descent segments detected by large-scale pedigree analysis
Eric Y. Durand, Nicholas Eriksson, Cory Y. McLean
(Submitted on 5 Nov 2013)
Analysis of genomic segments shared identical-by-descent (IBD) between individuals is fundamental to many genetic applications, but IBD detection accuracy in non-simulated data is largely unknown. Using 25,432 genotyped European individuals, and exploiting known familial relationships in 2,952 father-mother-child trios contained therein, we identify a false positive rate over 67% for short (2-4 centiMorgan) segments. We introduce a novel, computationally-efficient, haplotype-based metric that enables accurate IBD detection on population-scale datasets.
Link
arXiv:1311.1120 [q-bio.PE]
Reducing pervasive false positive identical-by-descent segments detected by large-scale pedigree analysis
Eric Y. Durand, Nicholas Eriksson, Cory Y. McLean
(Submitted on 5 Nov 2013)
Analysis of genomic segments shared identical-by-descent (IBD) between individuals is fundamental to many genetic applications, but IBD detection accuracy in non-simulated data is largely unknown. Using 25,432 genotyped European individuals, and exploiting known familial relationships in 2,952 father-mother-child trios contained therein, we identify a false positive rate over 67% for short (2-4 centiMorgan) segments. We introduce a novel, computationally-efficient, haplotype-based metric that enables accurate IBD detection on population-scale datasets.
Link
November 05, 2013
Population structure in Thailand

Insight into the Peopling of Mainland Southeast Asia from Thai Population Genetic Structure
Pongsakorn Wangkumhang et al.
There is considerable ethno-linguistic and genetic variation among human populations in Asia, although tracing the origins of this diversity is complicated by migration events. Thailand is at the center of Mainland Southeast Asia (MSEA), a region within Asia that has not been extensively studied. Genetic substructure may exist in the Thai population, since waves of migration from southern China throughout its recent history may have contributed to substantial gene flow. Autosomal SNP data were collated for 438,503 markers from 992 Thai individuals. Using the available self-reported regional origin, four Thai subpopulations genetically distinct from each other and from other Asian populations were resolved by Neighbor-Joining analysis using a 41,569 marker subset. Using an independent Principal Components-based unsupervised clustering approach, four major MSEA subpopulations were resolved in which regional bias was apparent. A major ancestry component was common to these MSEA subpopulations and distinguishes them from other Asian subpopulations. On the other hand, these MSEA subpopulations were admixed with other ancestries, in particular one shared with Chinese. Subpopulation clustering using only Thai individuals and the complete marker set resolved four subpopulations, which are distributed differently across Thailand. A Sino-Thai subpopulation was concentrated in the Central region of Thailand, although this constituted a minority in an otherwise diverse region. Among the most highly differentiated markers which distinguish the Thai subpopulations, several map to regions known to affect phenotypic traits such as skin pigmentation and susceptibility to common diseases. The subpopulation patterns elucidated have important implications for evolutionary and medical genetics. The subpopulation structure within Thailand may reflect the contributions of different migrants throughout the history of MSEA. The information will also be important for genetic association studies to account for population-structure confounding effects.
Link
European pigs replacing Near Eastern ones in Iron Age Israel

Scientific Reports 3, Article number: 3035 doi:10.1038/srep03035
Ancient DNA and Population Turnover in Southern Levantine Pigs- Signature of the Sea Peoples Migration?
Meirav Meiri et al.
Near Eastern wild boars possess a characteristic DNA signature. Unexpectedly, wild boars from Israel have the DNA sequences of European wild boars and domestic pigs. To understand how this anomaly evolved, we sequenced DNA from ancient and modern pigs from Israel. Pigs from Late Bronze Age (until ca. 1150 BCE) in Israel shared haplotypes of modern and ancient Near Eastern pigs. European haplotypes became dominant only during the Iron Age (ca. 900 BCE). This raises the possibility that European pigs were brought to the region by the Sea Peoples who migrated to the Levant at that time. Then, a complete genetic turnover took place, most likely because of repeated admixture between local and introduced European domestic pigs that went feral. Severe population bottlenecks likely accelerated this process. Introductions by humans have strongly affected the phylogeography of wild animals, and interpretations of phylogeography based on modern DNA alone should be taken with caution.
Link