
November 30, 2012
Using Genographic 2.0 data with DIYDodecad
I have released a converter for Genographic 2.0 data at the Dodecad blog. This will allow you to use DIYDodecad with your Genographic 2.0 raw data download.
November 29, 2012
Pinpointing Roma origins: Out of Northwestern India
Interestingly, besides H-M82, there has been recent evidence that R-Z93 might also represent a second founder haplogroup of the European Roma populations; it will be interesting to study it in the future in order to confirm the scenario presented in this new paper.
From the paper:
PLoS ONE 7(11): e48477. doi:10.1371/journal.pone.0048477
The Phylogeography of Y-Chromosome Haplogroup H1a1a-M82 Reveals the Likely Indian Origin of the European Romani Populations
Niraj Rai et al.
Linguistic and genetic studies on Roma populations inhabited in Europe have unequivocally traced these populations to the Indian subcontinent. However, the exact parental population group and time of the out-of-India dispersal have remained disputed. In the absence of archaeological records and with only scanty historical documentation of the Roma, comparative linguistic studies were the first to identify their Indian origin. Recently, molecular studies on the basis of disease-causing mutations and haploid DNA markers (i.e. mtDNA and Y-chromosome) supported the linguistic view. The presence of Indian-specific Y-chromosome haplogroup H1a1a-M82 and mtDNA haplogroups M5a1, M18 and M35b among Roma has corroborated that their South Asian origins and later admixture with Near Eastern and European populations. However, previous studies have left unanswered questions about the exact parental population groups in South Asia. Here we present a detailed phylogeographical study of Y-chromosomal haplogroup H1a1a-M82 in a data set of more than 10,000 global samples to discern a more precise ancestral source of European Romani populations. The phylogeographical patterns and diversity estimates indicate an early origin of this haplogroup in the Indian subcontinent and its further expansion to other regions. Tellingly, the short tandem repeat (STR) based network of H1a1a-M82 lineages displayed the closest connection of Romani haplotypes with the traditional scheduled caste and scheduled tribe population groups of northwestern India.
Link
From the paper:
This first genetic evidence of this nature allows us to develop a more detailed picture of the paternal genetic history of European Roma, revealing that the ancestors of present scheduled tribes and scheduled caste populations of northern India, traditionally referred to collectively as the Ḍoma, are the likely ancestral populations of modern European Roma. Our findings corroborate the hypothesized cognacy of the terms Rroma and Ḍoma and resolve the controversy about the Gangetic plain and the Punjab in favour of the northwestern portion of the diffuse widespread range of the Ḍoma ancestral population of northern India.A paper about Roma origins based on autosomal DNA is also apparently in the works, so it will be interesting to see how it might tie in with the Y-chromosome evidence.
PLoS ONE 7(11): e48477. doi:10.1371/journal.pone.0048477
The Phylogeography of Y-Chromosome Haplogroup H1a1a-M82 Reveals the Likely Indian Origin of the European Romani Populations
Niraj Rai et al.
Linguistic and genetic studies on Roma populations inhabited in Europe have unequivocally traced these populations to the Indian subcontinent. However, the exact parental population group and time of the out-of-India dispersal have remained disputed. In the absence of archaeological records and with only scanty historical documentation of the Roma, comparative linguistic studies were the first to identify their Indian origin. Recently, molecular studies on the basis of disease-causing mutations and haploid DNA markers (i.e. mtDNA and Y-chromosome) supported the linguistic view. The presence of Indian-specific Y-chromosome haplogroup H1a1a-M82 and mtDNA haplogroups M5a1, M18 and M35b among Roma has corroborated that their South Asian origins and later admixture with Near Eastern and European populations. However, previous studies have left unanswered questions about the exact parental population groups in South Asia. Here we present a detailed phylogeographical study of Y-chromosomal haplogroup H1a1a-M82 in a data set of more than 10,000 global samples to discern a more precise ancestral source of European Romani populations. The phylogeographical patterns and diversity estimates indicate an early origin of this haplogroup in the Indian subcontinent and its further expansion to other regions. Tellingly, the short tandem repeat (STR) based network of H1a1a-M82 lineages displayed the closest connection of Romani haplotypes with the traditional scheduled caste and scheduled tribe population groups of northwestern India.
Link
South Indian Y chromosomes (+ a little complaining about methods)
.png)
First of all, it is inconceivable to me how scientists can continue to use the 3x slower "evolutionary mutation rate" for their analyses of Y-chromosome ages on the basis of Y-STR markers. I have done my small part in my Y-STR series to show that this mutation rate is applicable only for a rather specific demographic history, and completely unsuitable to real growing human populations where Y-STR variance accumulates at close to the genealogical rate. And, my observations merely elaborated quantitatively what was already present in Zhivotovsky et al. (2006) but has been completely ignored since:
In simulations of a neutral process with average rate of increase m = 1, the number of surviving haplogroups rapidly decreased with time and corresponded well with the theory of mutant survival (Li 1955, p. 242), and the average size of the surviving haplogroups increased each generation by a value rapidly approaching 0.5 (data not shown), which agrees with asymptotic fraction of 2/t of haplotypes that survive at generation t (Athreya and Ney 1972, p. 19). The accumulated variance increased almost linearly (fig. 1), at a rate of increase about 0.00028 per generation; that is, the actual rate of accumulation microsatellite variation was about 3.6 times less than that predicted from the germ line mutation rate. This corresponds perfectly to the 3- to 4-fold difference observed between germ line and evolutionarily effective mutation rate.The issue is all but resolved in the amateur "genetic genealogy" community, but even professional geneticists often use either genealogical or evolutionary rate, or take an agnostic stance by reporting results based on both rates. To arrive at strong conclusions about a topic on the basis of a mutation rate that is, to say the least, controversial, without even acknowledging the existence of a controversy is unsatisfactory. Y-chromosome researchers ought to copy the attitude of those working with autosomal DNA, where a corresponding mutation rate controversy was not swept under the carpet, but acknowledged (e.g., in the recent Meyer et al. high-coverage Denisova paper), with the implications of the uncertainty during the present "transitional" period quantified in the form of wider confidence intervals.
This "mutation rate" issue notwithstanding, it was also recently shown that by Busby et al. that Y-STR based estimates have a dependence on the set of Y-STRs used, with markers exhibiting linear behavior across different time spans. This does not invalidate their use as molecular clocks, but highlights the need to not only select a bunch of Y-STRs, but also either (i) demonstrate that the selected set exhibits linear behavior for the time span of interest, or (ii) correct for deviations from linearity. Again, this type of modelling of microsatellite behavior was recently achieved for autosomal STRs by Sun et al. Note that such deviations result in a slower rate than the genealogical one, but the mechanism whereby this is produced is completely different than the one proposed by Zhivotovsky et al.: it is not drift in a non-growing (m=1) population that reduces the effective rate, but rather "saturation" of the mutation process, whereby the variance at fast-mutating markers grows sub-linearly with time, because of physical constraints on their possible range of values.
I don't hope that Y-STR based age estimation will have much to offer in the coming years. But the third set of the 1000 Genomes Project is on its way, and this will include a variety of South Asian samples. Very soon we will be in a good position to study the time depth of common ancestry between e.g., European and South Asian Y-chromosomes within various haplogroups using point mutations, and these are not plagued by many of the problems associated with Y-STR variation and its interpretation.
Finally, I can't help but notice that this paper has not acknowledged the tremendous progress in resolving the Y chromosome phylogeny done by non-academic researchers. With the current state of our knowledge, the claim that haplogroup R1a1 is "autochthonous" in India is not tenable. Even if one discounts all the evidence made by SNP discoveries in the commercial testing world (and why should they?), finer-scale structure within this haplogroup has now been officially published and appears to be inconsistent with a South Asian origin of this haplogroup.
Certainly, not all is resolved; for example, the representation of tribal populations in commercial DNA testing is almost non-existent, and a sampling of their Y-SNP diversity is urgently needed. A very useful paradigm of research is that of recent work on the most basal clade of the Y-chromosome phylogeny (A00) in which the identification of very unique Y-chromosomes by genetic genealogists was combined with academic samples of "indigenous" peoples to produce new knowledge.
Much of population genetic research will benefit from such consilience between academics and amateurs. This is not an idle hope, but a recognition that this field is one in which the public not only has a substantial interest but can also do something about it. Many might be interested in Mars exploration, but without Elon Musk's bank account, most are consigned to being consumers of information about the Red Planet. Hopefully, better ways of combining the efforts of research scientists and the educated public can be identified and used in the near future.
PLoS ONE 7(11): e50269. doi:10.1371/journal.pone.0050269
Population Differentiation of Southern Indian Male Lineages Correlates with Agricultural Expansions Predating the Caste System
GaneshPrasad ArunKumar et al.
Previous studies that pooled Indian populations from a wide variety of geographical locations, have obtained contradictory conclusions about the processes of the establishment of the Varna caste system and its genetic impact on the origins and demographic histories of Indian populations. To further investigate these questions we took advantage that both Y chromosome and caste designation are paternally inherited, and genotyped 1,680 Y chromosomes representing 12 tribal and 19 non-tribal (caste) endogamous populations from the predominantly Dravidian-speaking Tamil Nadu state in the southernmost part of India. Tribes and castes were both characterized by an overwhelming proportion of putatively Indian autochthonous Y-chromosomal haplogroups (H-M69, F-M89, R1a1-M17, L1-M27, R2-M124, and C5-M356; 81% combined) with a shared genetic heritage dating back to the late Pleistocene (10–30 Kya), suggesting that more recent Holocene migrations from western Eurasia contributed less than 20% of the male lineages. We found strong evidence for genetic structure, associated primarily with the current mode of subsistence. Coalescence analysis suggested that the social stratification was established 4–6 Kya and there was little admixture during the last 3 Kya, implying a minimal genetic impact of the Varna (caste) system from the historically-documented Brahmin migrations into the area. In contrast, the overall Y-chromosomal patterns, the time depth of population diversifications and the period of differentiation were best explained by the emergence of agricultural technology in South Asia. These results highlight the utility of detailed local genetic studies within India, without prior assumptions about the importance of Varna rank status for population grouping, to obtain new insights into the relative influences of past demographic events for the population structure of the whole of modern India.
Link
November 28, 2012
Paleoamerican Odyssey conference, ~a year from now
Here is a list of abstracts from a conference that will take place in October 2013. A small sampling of interesting titles:
There is also the issue of the Solutrean hypothesis; if early North Americans had European ancestors, and the early population was diluted by subsequent population movements from Asia, this ought to show up. Additionally, there is the hypothesis of a common North Eurasian ancestry affecting both Europe and Amerindians, which would predict that the Clovis individual would be an early descendant possessing that type of ancestry.
In about a year we might know much more about the identity of early New World populations, and, by implication, adapt our views about the settling of the Old World itself.
- Yana RHS site, earliest occupation of Siberia
- Late Pleistocene Siberia: Setting the Stage for the Peopling of the Americas
- Three Stage Colonization Model for the Peopling of the Americas
- The Younger Dryas Boundary (YDB) Cosmic Impact Hypothesis, 12.9 ka: A Review
- Bioarchaeological Biographies of Ancient Americans
- Paisley Caves: 14,500 Years of Human Occupations in the Northern Great Basin
- The Mammoth Steppe Hypothesis: The Mid Wisconsin (OIS 3) Peopling of the Americas
- North America Before Clovis: Variance in Temporal/Spatial Cultural Patterns, 24,000 to 13,000 BP
On the archaeogenetics side, an intriguing abstract of Eske Willerslev's talk:
A Genomic Sequence of a Clovis Individual
Eske Willerslev
The Clovis complex is by some scientists considered being the oldest unequivocal evidence of humans in the Americas, dating between ca. 11,050 to 10,800 14C yr B.P. Only one human skeleton has been directly AMS dated to Clovis age and found associated with Clovis technology namely the Anzick human remains from Montana. We are currently sequencing the nuclear and mitochondrial genome from this human skeleton in order to address the origins and descendents of Clovis. I will present the results obtained by our international consortium.In terms of the "three-migration" model, Clovis ought to be "First American". But, there is evidence that at least archaeologically Clovis had company and predecessors, so it will be interesting to see how closely the sample will match our expectation of what "First American" DNA looked like.
There is also the issue of the Solutrean hypothesis; if early North Americans had European ancestors, and the early population was diluted by subsequent population movements from Asia, this ought to show up. Additionally, there is the hypothesis of a common North Eurasian ancestry affecting both Europe and Amerindians, which would predict that the Clovis individual would be an early descendant possessing that type of ancestry.
In about a year we might know much more about the identity of early New World populations, and, by implication, adapt our views about the settling of the Old World itself.
November 27, 2012
Ancestry Mapper (Magalhães et al. 2012)
The idea of Ancestry Mapper is fairly simple: each individual is represented as a vector of similarity to a fixed number of a priori chosen reference populations. These vectors can then be processed (e.g., with clustering) as any other type of high-dimensional data (e.g., PC co-ordinates).
.png)
This was produced by applying PAM clustering to AMids. I don't think that this is a better way to do clustering than PCA/MDS+MCLUST, both because "partition around medoids" is a less expressive model than the suite of models that MCLUST may consider and choose from, and also because the AMids assume a priori assignment of individuals to populations, which is not necessary for the "Galore" approach that uses MDS/PCA for dimensionality reduction of individuals and is agnostic about their population labels. In any case, it is useful to know that with both a different dimensionality reduction method and a different clustering algorithm, a large number of meaningful clusters can be inferred.
PLoS ONE 7(11): e49438. doi:10.1371/journal.pone.0049438
HGDP and HapMap Analysis by Ancestry Mapper Reveals Local and Global Population Relationships
Knowledge of human origins, migrations, and expansions is greatly enhanced by the availability of large datasets of genetic information from different populations and by the development of bioinformatic tools used to analyze the data. We present Ancestry Mapper, which we believe improves on existing methods, for the assignment of genetic ancestry to an individual and to study the relationships between local and global populations. The principle function of the method, named Ancestry Mapper, is to give each individual analyzed a genetic identifier, made up of just 51 genetic coordinates, that corresponds to its relationship to the HGDP reference population. As a consequence, the Ancestry Mapper Id (AMid) has intrinsic biological meaning and provides a tool to measure similarity between world populations. We applied Ancestry Mapper to a dataset comprised of the HGDP and HapMap data. The results show distinctions at the continental level, while simultaneously giving details at the population level. We clustered AMids of HGDP/HapMap and observe a recapitulation of human migrations: for a small number of clusters, individuals are grouped according to continental origins; for a larger number of clusters, regional and population distinctions are evident. Calculating distances between AMids allows us to infer ancestry. The number of coordinates is expandable, increasing the power of Ancestry Mapper. An R package called Ancestry Mapper is available to apply this method to any high density genomic data set.
Link
The following figure should appear familiar to readers familiar with my MDS/MCLUST "Clusters Galore" methodology:
.png)
This was produced by applying PAM clustering to AMids. I don't think that this is a better way to do clustering than PCA/MDS+MCLUST, both because "partition around medoids" is a less expressive model than the suite of models that MCLUST may consider and choose from, and also because the AMids assume a priori assignment of individuals to populations, which is not necessary for the "Galore" approach that uses MDS/PCA for dimensionality reduction of individuals and is agnostic about their population labels. In any case, it is useful to know that with both a different dimensionality reduction method and a different clustering algorithm, a large number of meaningful clusters can be inferred.
PLoS ONE 7(11): e49438. doi:10.1371/journal.pone.0049438
HGDP and HapMap Analysis by Ancestry Mapper Reveals Local and Global Population Relationships
Knowledge of human origins, migrations, and expansions is greatly enhanced by the availability of large datasets of genetic information from different populations and by the development of bioinformatic tools used to analyze the data. We present Ancestry Mapper, which we believe improves on existing methods, for the assignment of genetic ancestry to an individual and to study the relationships between local and global populations. The principle function of the method, named Ancestry Mapper, is to give each individual analyzed a genetic identifier, made up of just 51 genetic coordinates, that corresponds to its relationship to the HGDP reference population. As a consequence, the Ancestry Mapper Id (AMid) has intrinsic biological meaning and provides a tool to measure similarity between world populations. We applied Ancestry Mapper to a dataset comprised of the HGDP and HapMap data. The results show distinctions at the continental level, while simultaneously giving details at the population level. We clustered AMids of HGDP/HapMap and observe a recapitulation of human migrations: for a small number of clusters, individuals are grouped according to continental origins; for a larger number of clusters, regional and population distinctions are evident. Calculating distances between AMids allows us to infer ancestry. The number of coordinates is expandable, increasing the power of Ancestry Mapper. An R package called Ancestry Mapper is available to apply this method to any high density genomic data set.
Link
Skull trauma in Neolithic Scandinavia
I would be interested to know how this Neolithic sample might differ from more recent ones. My limited understanding suggests that between-male violence often has a signalling component whereby an individual's or group's dominance over another is asserted, so the fight often does not go all the way to death, but only until the status quo is manifested by the controlling party or toppled by a challenger.
This type of "signalling" aspect of violent behavior does not apply to male-to-female violence because of the physical strength inequality between the sexes. Indeed, as with violence towards children or the elderly, male-to-female violence may have a "reverse signalling" effect, because it suggests that the perpetrator is unable to fight with "the strong" and is only able to assert physical dominance in "easy fights". On the other hand, such "easy fights" might be more abundant if perpetrators tend to enter fights they can win.
Fight-to-the-death, on the other hand, may occur either by accident (e.g., when the aim is to assert dominance, but the killer underestimates the tolerance of the victim), or by intent (when the aim is physical annihilation, either because reconciliation with the victim is perceived to be impossible, or because the victim's death may help keep other challengers in check).
There may be lots to learn about gender roles and social hierarchy from large palaeoanthropological samples. For example, how much did ideology affect secular patterns of interpersonal violence, and how much did changes in weapon technology (e.g., from Neolithic to Bronze, Iron, and more recently firearms).
Am J Phys Anthropol DOI: 10.1002/ajpa.22192
Patterns of violence-related skull trauma in neolithic southern scandinavia
Linda Fibiger et al.
This article examines evidence for violence as reflected in skull injuries in 378 individuals from Neolithic Denmark and Sweden (3,900–1,700 BC). It is the first large-scale crossregional study of skull trauma in southern Scandinavia, documenting skeletal evidence of violence at a population level. We also investigate the widely assumed hypothesis that Neolithic violence is male-dominated and results in primarily male injuries and fatalities. Considering crude prevalence and prevalence for individual bones of the skull allows for a more comprehensive understanding of interpersonal violence in the region, which is characterized by endemic levels of mostly nonlethal violence that affected both men and women. Crude prevalence for skull trauma reaches 9.4% in the Swedish and 16.9% in the Danish sample, whereas element-based prevalence varies between 6.2% for the right frontal and 0.6% for the left maxilla, with higher figures in the Danish sample. Significantly more males are affected by healed injuries but perimortem injuries affect males and females equally. These results suggest habitual male involvement in nonfatal violence but similar risks for both sexes for sustaining fatal injuries. In the Danish sample, a bias toward front and left-side injuries and right-side injuries in females support this scenario of differential involvement in habitual interpersonal violence, suggesting gendered differences in active engagement in conflict. It highlights the importance of large-scale studies for investigating the scale and context of violence in early agricultural societies, and the existence of varied regional patterns for overall injury prevalence as well as gendered differences in violence-related injuries.
Link
This type of "signalling" aspect of violent behavior does not apply to male-to-female violence because of the physical strength inequality between the sexes. Indeed, as with violence towards children or the elderly, male-to-female violence may have a "reverse signalling" effect, because it suggests that the perpetrator is unable to fight with "the strong" and is only able to assert physical dominance in "easy fights". On the other hand, such "easy fights" might be more abundant if perpetrators tend to enter fights they can win.
Fight-to-the-death, on the other hand, may occur either by accident (e.g., when the aim is to assert dominance, but the killer underestimates the tolerance of the victim), or by intent (when the aim is physical annihilation, either because reconciliation with the victim is perceived to be impossible, or because the victim's death may help keep other challengers in check).
There may be lots to learn about gender roles and social hierarchy from large palaeoanthropological samples. For example, how much did ideology affect secular patterns of interpersonal violence, and how much did changes in weapon technology (e.g., from Neolithic to Bronze, Iron, and more recently firearms).
Am J Phys Anthropol DOI: 10.1002/ajpa.22192
Patterns of violence-related skull trauma in neolithic southern scandinavia
Linda Fibiger et al.
This article examines evidence for violence as reflected in skull injuries in 378 individuals from Neolithic Denmark and Sweden (3,900–1,700 BC). It is the first large-scale crossregional study of skull trauma in southern Scandinavia, documenting skeletal evidence of violence at a population level. We also investigate the widely assumed hypothesis that Neolithic violence is male-dominated and results in primarily male injuries and fatalities. Considering crude prevalence and prevalence for individual bones of the skull allows for a more comprehensive understanding of interpersonal violence in the region, which is characterized by endemic levels of mostly nonlethal violence that affected both men and women. Crude prevalence for skull trauma reaches 9.4% in the Swedish and 16.9% in the Danish sample, whereas element-based prevalence varies between 6.2% for the right frontal and 0.6% for the left maxilla, with higher figures in the Danish sample. Significantly more males are affected by healed injuries but perimortem injuries affect males and females equally. These results suggest habitual male involvement in nonfatal violence but similar risks for both sexes for sustaining fatal injuries. In the Danish sample, a bias toward front and left-side injuries and right-side injuries in females support this scenario of differential involvement in habitual interpersonal violence, suggesting gendered differences in active engagement in conflict. It highlights the importance of large-scale studies for investigating the scale and context of violence in early agricultural societies, and the existence of varied regional patterns for overall injury prevalence as well as gendered differences in violence-related injuries.
Link
November 26, 2012
Medieval signal of Swedish (?) admixture in Finland
I took the FIN (Finnish), GBR (British), and CDX (Chinese Dai) samples of the 1000 Genomes Project, each of which has a sample size of 100 in order to investigate the signal of East-West Eurasian admixture in Finns. While neither Britons nor Dai could be imagine of having contributed to Finns directly, they ought to make useful proxies of a NW European population lacking recent East Eurasian ancestry, and an East Eurasian population lacking recent West Eurasian ancestry respectively.
In the following, I will assume a generation length of 29 years and a sample birthyear of 1980 as in previous experiments.
First, the 1-reference analysis of FIN using GBR produced an admixture proportion lower bound of 37.4 +/- 5.1 percent.
The corresponding analysis of FIN using CDX produced an admixture proportion lower bound of 4.4 +/- 1.0 percent.
The 2-ref admixture test with {GBR,CDX} reported success:
In the following, I will assume a generation length of 29 years and a sample birthyear of 1980 as in previous experiments.
First, the 1-reference analysis of FIN using GBR produced an admixture proportion lower bound of 37.4 +/- 5.1 percent.
The corresponding analysis of FIN using CDX produced an admixture proportion lower bound of 4.4 +/- 1.0 percent.
The 2-ref admixture test with {GBR,CDX} reported success:
Test SUCCEEDS (z=2.76, p=0.0057) for FIN with {GBR, CDX} weights
But, the decay rates were inconsistent, a situation which might occur when major admixture from different sources took place at different times. In particular, the one using CDX corresponded to 65.57 +/- 8.36 generations, and the one using GBR to 25.48 +/- 4.93 generations.
In calendar dates, Finns are estimated to have mixed with an East Eurasian CDX-like population between 170BC-320AD and with a NW European GBR-like population between 1100-1380AD.
The central date of the latter estimate is 1,240AD, which corresponds quite closely to the beginning of Swedish rule and is in the middle of the 13th. century, between the time when Finland was initially claimed for western Christendom (12th c.) and the time when the conflict between Sweden and Russia was settled (14th c.).
Irish Travellers are Irish
Am J Phys Anthropol DOI: 10.1002/ajpa.22191
Genetic drift and the population history of the Irish travellers
John H. Relethford, Michael H. Crawford
Abstract
The Irish Travellers are an itinerant group in Ireland that has been socially isolated. Two hypotheses have been proposed concerning the genetic origin of the Travellers: (1) they are genetically related to Roma populations in Europe that share a nomadic lifestyle or (2) they are of Irish origin, and genetic differences from the rest of Ireland reflect genetic drift. These hypotheses were tested using data on 33 alleles from 12 red blood cell polymorphism loci. Comparison with other European, Roma, and Indian populations shows that the Travellers are genetically distinct from the Roma and Indian populations and most genetically similar to Ireland, in agreement with earlier genetic analyses of the Travellers. However, the Travellers are still genetically distinct from other Irish populations, which could reflect some external gene flow and/or the action of genetic drift in a small group that was descended from a small number of founders. In order to test the drift hypothesis, we analyzed genetic distances comparing the Travellers to four geographic regions in Ireland. These distances were then compared with adjusted distances that account for differential genetic drift using a method developed by Relethford (Hum Biol 68 (1996) 29–44). The unadjusted distances show the genetic distinctiveness of the Travellers. After adjustment for the expected effects of genetic drift, the Travellers are equidistant from the other Irish samples, showing their Irish origins and population history. The observed genetic differences are thus a reflection of genetic drift, and there is no evidence of any external gene flow.
Link
Genetic drift and the population history of the Irish travellers
John H. Relethford, Michael H. Crawford
Abstract
The Irish Travellers are an itinerant group in Ireland that has been socially isolated. Two hypotheses have been proposed concerning the genetic origin of the Travellers: (1) they are genetically related to Roma populations in Europe that share a nomadic lifestyle or (2) they are of Irish origin, and genetic differences from the rest of Ireland reflect genetic drift. These hypotheses were tested using data on 33 alleles from 12 red blood cell polymorphism loci. Comparison with other European, Roma, and Indian populations shows that the Travellers are genetically distinct from the Roma and Indian populations and most genetically similar to Ireland, in agreement with earlier genetic analyses of the Travellers. However, the Travellers are still genetically distinct from other Irish populations, which could reflect some external gene flow and/or the action of genetic drift in a small group that was descended from a small number of founders. In order to test the drift hypothesis, we analyzed genetic distances comparing the Travellers to four geographic regions in Ireland. These distances were then compared with adjusted distances that account for differential genetic drift using a method developed by Relethford (Hum Biol 68 (1996) 29–44). The unadjusted distances show the genetic distinctiveness of the Travellers. After adjustment for the expected effects of genetic drift, the Travellers are equidistant from the other Irish samples, showing their Irish origins and population history. The observed genetic differences are thus a reflection of genetic drift, and there is no evidence of any external gene flow.
Link
LAMP-LD paper and software
On a similar topic as the recent MULTIMIX software, this paper describes the performance of LAMP-LD software on Latinos with ancestry from Europe, Africa, and the Americas. The software can be obtained from this site.
From the paper itself, this figure highlights a problem I have previously identified:

In this experiment, the authors "European-ized" East Asian reference panels by introducing TSI (Tuscan) segments into them. From the paper:
In the case of Native American admixture, the occurrence of European segments in the reference panels is a problem, because we can be fairly sure that prior to 1492 there was no recent European ancestry in the Americas.
But, the problem also arises in other cases where this is less certain, for example the arrival of East Eurasian ancestry via Uralic and Turkic speakers from Siberia and Central Asia and into West Eurasia. In that case, we cannot be entirely certain whether the presence of European haplotypes in reference populations (e.g., present-day Siberians/Central Asians) is due to post- or pre-migration contact in the eastern source areas.
To make my observation clearer: suppose that an eastern population X, contributes to a European population Y. We can then estimate how much "X ancestry" population Y has absorbed. But, if X today is "more East Asian" than X when it contributed to Y, then the proportion of admixture will be underestimated, and in the converse case it will be overestimated.
This was made evident in my recent analysis of Turks where substantially different admixture estimates was obtained using different eastern populations. The evidence of that analysis suggests that major admixture occurred in Central Asia after it did in Anatolia.
Bioinformatics (2012) 28 (10): 1359-1367. doi: 10.1093/bioinformatics/bts144
Fast and accurate inference of local ancestry in Latino populations
Yael Baran et al.
Motivation: It is becoming increasingly evident that the analysis of genotype data from recently admixed populations is providing important insights into medical genetics and population history. Such analyses have been used to identify novel disease loci, to understand recombination rate variation and to detect recent selection events. The utility of such studies crucially depends on accurate and unbiased estimation of the ancestry at every genomic locus in recently admixed populations. Although various methods have been proposed and shown to be extremely accurate in two-way admixtures (e.g. African Americans), only a few approaches have been proposed and thoroughly benchmarked on multi-way admixtures (e.g. Latino populations of the Americas).
Results: To address these challenges we introduce here methods for local ancestry inference which leverage the structure of linkage disequilibrium in the ancestral population (LAMP-LD), and incorporate the constraint of Mendelian segregation when inferring local ancestry in nuclear family trios (LAMP-HAP). Our algorithms uniquely combine hidden Markov models (HMMs) of haplotype diversity within a novel window-based framework to achieve superior accuracy as compared with published methods. Further, unlike previous methods, the structure of our HMM does not depend on the number of reference haplotypes but on a fixed constant, and it is thereby capable of utilizing large datasets while remaining highly efficient and robust to over-fitting. Through simulations and analysis of real data from 489 nuclear trio families from the mainland US, Puerto Rico and Mexico, we demonstrate that our methods achieve superior accuracy compared with published methods for local ancestry inference in Latinos.
Availability: http://lamp.icsi.berkeley.edu/lamp/lampld/
Link
From the paper itself, this figure highlights a problem I have previously identified:

In this experiment, the authors "European-ized" East Asian reference panels by introducing TSI (Tuscan) segments into them. From the paper:
Current day Native American haplotypes used as proxy for the Native American component of Latinos are presumed to contain European gene flow. In order to test the effect of this phenomenon on ancestry inference, we introduced TSI segments into the Asian haplotypes of a reference set composed of 117 CEU, 169 (CHB+CHD) and 115 YRI haplotypes. We performed 10 experiments, in each choosing at random a 5 Mb region along the chromosome, and replacing a percentage of the (CHB+CHD) haplotypes with TSI haplotypes along the chosen region.
We observed that the typical effect of increasing the number of TSI segments present in the Native American reference panels is an increase in the estimated proportion of the Native American ancestry along the modified region, at the expense of the estimated European proportion.
In the case of Native American admixture, the occurrence of European segments in the reference panels is a problem, because we can be fairly sure that prior to 1492 there was no recent European ancestry in the Americas.
But, the problem also arises in other cases where this is less certain, for example the arrival of East Eurasian ancestry via Uralic and Turkic speakers from Siberia and Central Asia and into West Eurasia. In that case, we cannot be entirely certain whether the presence of European haplotypes in reference populations (e.g., present-day Siberians/Central Asians) is due to post- or pre-migration contact in the eastern source areas.
To make my observation clearer: suppose that an eastern population X, contributes to a European population Y. We can then estimate how much "X ancestry" population Y has absorbed. But, if X today is "more East Asian" than X when it contributed to Y, then the proportion of admixture will be underestimated, and in the converse case it will be overestimated.
This was made evident in my recent analysis of Turks where substantially different admixture estimates was obtained using different eastern populations. The evidence of that analysis suggests that major admixture occurred in Central Asia after it did in Anatolia.
Bioinformatics (2012) 28 (10): 1359-1367. doi: 10.1093/bioinformatics/bts144
Fast and accurate inference of local ancestry in Latino populations
Yael Baran et al.
Motivation: It is becoming increasingly evident that the analysis of genotype data from recently admixed populations is providing important insights into medical genetics and population history. Such analyses have been used to identify novel disease loci, to understand recombination rate variation and to detect recent selection events. The utility of such studies crucially depends on accurate and unbiased estimation of the ancestry at every genomic locus in recently admixed populations. Although various methods have been proposed and shown to be extremely accurate in two-way admixtures (e.g. African Americans), only a few approaches have been proposed and thoroughly benchmarked on multi-way admixtures (e.g. Latino populations of the Americas).
Results: To address these challenges we introduce here methods for local ancestry inference which leverage the structure of linkage disequilibrium in the ancestral population (LAMP-LD), and incorporate the constraint of Mendelian segregation when inferring local ancestry in nuclear family trios (LAMP-HAP). Our algorithms uniquely combine hidden Markov models (HMMs) of haplotype diversity within a novel window-based framework to achieve superior accuracy as compared with published methods. Further, unlike previous methods, the structure of our HMM does not depend on the number of reference haplotypes but on a fixed constant, and it is thereby capable of utilizing large datasets while remaining highly efficient and robust to over-fitting. Through simulations and analysis of real data from 489 nuclear trio families from the mainland US, Puerto Rico and Mexico, we demonstrate that our methods achieve superior accuracy compared with published methods for local ancestry inference in Latinos.
Availability: http://lamp.icsi.berkeley.edu/lamp/lampld/
Link
November 24, 2012
Lack of β thalassemia mutations in Minoan Cretans
Blood Cells, Molecules, and Diseases
Volume 48, Issue 1, 15 January 2012, Pages 7–10
A search for β thalassemia mutations in 4000 year old ancient DNAs of Minoan Cretans
Jeffery R. Hughey et al.
Ancient DNA methodologies can be applied in the investigation of the genetics of extinct populations. A search for beta thalassemia mutations was performed on 49 Minoan individuals from the Bronze Age who were living in the island of Crete approximately 4000 Years Before Present (YBP). Standard precautionary measures were employed in the laboratory to ensure authenticity of the DNA extracted from the ancient bones, resulting in the successful analysis of DNA of 24 Minoans. DNA sequencing focused on the Intervening Sequence 1 (IVS-1) of the beta globin gene and its splicing junctions. 63% of the thalassemia mutations observed among modern Cretans reside in beta IVS-1. None of the Minoan individuals carried one of the IVS-1 mutations known to cause beta thalassemia; however, only one was expected to be observed if the average frequency of beta thalassemia heterozygotes in the Minoan population was the same with that of modern day Cretans (7.6%). One individual contained a C to G substitution in position 91 of the IVS-1, located 40 bp 5′ to the intron 1/exon 2 junction. Functional studies indicated that the mutation did not affect mRNA splicing or stability, and most likely represented an innocent single nucleotide polymorphism.
Link
A search for β thalassemia mutations in 4000 year old ancient DNAs of Minoan Cretans
Jeffery R. Hughey et al.
Ancient DNA methodologies can be applied in the investigation of the genetics of extinct populations. A search for beta thalassemia mutations was performed on 49 Minoan individuals from the Bronze Age who were living in the island of Crete approximately 4000 Years Before Present (YBP). Standard precautionary measures were employed in the laboratory to ensure authenticity of the DNA extracted from the ancient bones, resulting in the successful analysis of DNA of 24 Minoans. DNA sequencing focused on the Intervening Sequence 1 (IVS-1) of the beta globin gene and its splicing junctions. 63% of the thalassemia mutations observed among modern Cretans reside in beta IVS-1. None of the Minoan individuals carried one of the IVS-1 mutations known to cause beta thalassemia; however, only one was expected to be observed if the average frequency of beta thalassemia heterozygotes in the Minoan population was the same with that of modern day Cretans (7.6%). One individual contained a C to G substitution in position 91 of the IVS-1, located 40 bp 5′ to the intron 1/exon 2 junction. Functional studies indicated that the mutation did not affect mRNA splicing or stability, and most likely represented an innocent single nucleotide polymorphism.
Link
Assessment of Totonac and Bolivian samples using 'globe13'
I was on the lookout for some Affy 6.0 samples recently, and I discovered the data of the recent Watkins et al. (2012) paper, so I decided to run them through my globe13 calculator. A total of 49,233 SNPs were in common between that and my globe13 set, which is not much, but ought to be sufficient to discover the main features of these two population samples.

It appears that both samples are mainly "Amerindian", with the Bolivian sample having some more European admixture than the Totonac one.
Here are the population portraits, clearly showing that the "European" admixture in Bolivians comes from a subset of individuals.

 For comparison, here are the ADMIXTURE results from the original paper that appear quite similar to my own. (Note that the individual ordering is probably not the same as my own):
For comparison, here are the ADMIXTURE results from the original paper that appear quite similar to my own. (Note that the individual ordering is probably not the same as my own):

The Mediterranean/North_European ratio of my own analysis suggests the likely "southern" (probably Spanish) origin of the European admixture in these populations.
UPDATE:
I also combined the two Amerindian populations with HGDP Karitiana, Sardinian, and French to calculate f3-statistics. Here are the significant ones:

So, admixture in the Bolivian sample is confirmed, while in the Totonac one it is not. I do think it's possible that the Totonac might have a little European admixture though which might be masked by their history of drift. Also notice the evidence for admixture in the French using all three Amerindian samples, with lowest f3(French; Amerindian, Sardinian) using the Karitiana reference.
It appears that both samples are mainly "Amerindian", with the Bolivian sample having some more European admixture than the Totonac one.
Here are the population portraits, clearly showing that the "European" admixture in Bolivians comes from a subset of individuals.



The Mediterranean/North_European ratio of my own analysis suggests the likely "southern" (probably Spanish) origin of the European admixture in these populations.
UPDATE:
I also combined the two Amerindian populations with HGDP Karitiana, Sardinian, and French to calculate f3-statistics. Here are the significant ones:
So, admixture in the Bolivian sample is confirmed, while in the Totonac one it is not. I do think it's possible that the Totonac might have a little European admixture though which might be masked by their history of drift. Also notice the evidence for admixture in the French using all three Amerindian samples, with lowest f3(French; Amerindian, Sardinian) using the Karitiana reference.
November 23, 2012
The comings and goings of Near Eastern and European domestic pigs (Ottoni et al. 2012)
This is an excellent paper whose findings re: pig domestication seem to parallel many of my own observations regarding the flow of human populations. It is open access, so you can read it for yourselves, but the following figure illustrates the situation admirably:

The left-right arrangement of the columns corresponds to a west-east longitude across West Asia. It can be easily seen that some of the early domestic samples (yellow, bottom row) are concentrated in the west (Y1 haplotype), while others (blue, Arm1T) in the east.
Neolithic European samples possessed the Y1 haplotype, but lacked the Arm1T one. So, the authors conclude that:
In Europe itself, the early Near Eastern domestic pigs were replaced by European ones:
In any case, the interesting thing is that pigs carrying the "European" haplotype went the other way, crossing from Europe to Asia. The beginning of this process seems to have occurred in the Middle Bronze Age:
The beautiful temporal transect presented in the Figure may also prove useful for students of ancient human DNA. I'd love to see how humans living close to sites #14-16, dominated by Arm1T haplotypes throughout history might differ from those of Neolithic West Anatolia, and whether the "mixed" Iron Age sample from Lidar Höyük shows evidence of the arrival of European-like human populations to accompany the European pigs.
Mol Biol Evol (2012) doi: 10.1093/molbev/mss261
Pig domestication and human-mediated dispersal in western Eurasia revealed through ancient DNA and geometric morphometrics
Claudio Ottoni et al.
Zooarcheological evidence suggests that pigs were domesticated in Southwest Asia ∼8,500 BC. They then spread across the Middle and Near East and westward into Europe alongside early agriculturalists. European pigs were either domesticated independently or appeared so as a result of admixture between introduced pigs and European wild boar. These pigs not only replaced those with Near Eastern signatures in Europe, they subsequently also replaced indigenous domestic pigs in the Near East. The specific details of these processes, however, remain unknown. To address questions related to early pig domestication, dispersal, and turnover in the Near East, we analyzed ancient mitochondrial DNA and dental geometric morphometric variation in 393 ancient pig specimens representing 48 archeological sites (from the Pre-Pottery Neolithic to the Medieval period) from Armenia, Cyprus, Georgia, Iran, Syria and Turkey. Our results firstly reveal the genetic signature of early domestic pigs in Eastern Turkey. We also demonstrate that these early pigs differed genetically from those in western Anatolia that were introduced to Europe during the Neolithic expansion. In addition, we present a significantly more refined chronology for the introduction of European domestic pigs into Asia Minor that took place during the Bronze Age, nearly 1,000 years earlier than previously detected. By the 5th century AD, European signatures completely replaced the endemic lineages possibly coinciding with the demographic and societal changes during the Anatolian Bronze and Iron Ages.
Link

The left-right arrangement of the columns corresponds to a west-east longitude across West Asia. It can be easily seen that some of the early domestic samples (yellow, bottom row) are concentrated in the west (Y1 haplotype), while others (blue, Arm1T) in the east.
Neolithic European samples possessed the Y1 haplotype, but lacked the Arm1T one. So, the authors conclude that:
The ancient Anatolian data presented here reveal that both wild and possibly domestic Neolithic pigs (identified using traditional metrics) possessed Y1 haplotypes ... The presence of these lineages corroborates the supposition that the earliest domestic pigs in Europe originated from populations originally domesticated in the Near East, conclusively linking the Neolithization of Europe with Neolithic cultures of western Anatolia (Larson et al. 2007a; Haak et al. 2010).I have repeatedly highlighted the "puzzle" of the early European Neolithic: the signature Y-haplogroup G2a was unaccompanied by other common Near Eastern lineages, and the modal "West Asian" ancestral component in present-day West Asian populations seems to have been absent in early Neolithic samples, which were dominated by a "Sardinian-like" population. I have argued that this meant that the European Neolithic was drawn from a limited founder source that was more "Mediterranean/Southern" autosomally than "West Asian", at least in terms of the components identified by the Dodecad Project.
In Europe itself, the early Near Eastern domestic pigs were replaced by European ones:
Ancient DNA extracted from early Neolithic domestic pigs in Europe resolved this paradox by demonstrating that early domestic pigs in the Balkans and central Europe shared haplotypes with modern Near Eastern wild boar (Larson et al. 2007a). The absence of Near Eastern haplotypes in pre-Neolithic European wild boar suggested that early domestic pigs in Europe must have been introduced from the Near East by the mid 6th millennium BC before spreading to the Paris basin by the early 4th millennium BC (Larson et al. 2007a).
By 3,900 BC, however, virtually all domestic pigs in Europe possessed haplotypes from an indigenous European domestication process (Larson et al. 2007a) only found in European wild boar. This genetic turnover may have resulted from the accumulated introgression of local female wild boar into imported domestic stocks, or from an indigenous European domestication process (Larson et al. 2007a).We have seen that early Neolithic domestic pigs came from Western Anatolia, but apparently these did not last, but were replaced in Europe by pigs carrying mtDNA of European wild boar. An additional possibility is that the European wild boar were better adapted to local conditions in Europe, so the stock of European farmers gradually became "local" due to artificial/natural selection favoring the local "European" type. It might also be that in accordance with Bergmann's rule, European-descended pigs were simply bigger, and thus more economically productive.
In any case, the interesting thing is that pigs carrying the "European" haplotype went the other way, crossing from Europe to Asia. The beginning of this process seems to have occurred in the Middle Bronze Age:
The temporal and geographic distribution of genetic haplotypes presented in our study demonstrates that the first AMS dated pig with European ancestry (haplotype A) appeared almost 1,000 years earlier than the Armenian samples in a Late Bronze Age context (~1,600-1,440 BC) at Lidar Höyük (fig. 1). An even earlier Middle Bronze Age specimen from the same site also possessed a European signature, but a directI have written how increased mobility and long-range networks associated with the new metallurgical class facilitated commerce during the Bronze Age. The authors suggest the possibility of Minoan-Mycenaean/Hittite involvement during the Bronze Age, which are certainly plausible conduits for European pigs to have crossed the Aegean at this time. But, as you can see from the figure, the "European" pigs are still outliers during the Middle and Bronze Ages, but become common in the Iron Age sample from Lidar Höyük, and eventually replacing local types throughout Anatolia and Armenia, but, apparently, not Iran:
AMS date for this specimen could not be obtained.
The frequency of pigs with European ancestry increased rapidly from the 12th century BC, and by the 5th century AD domestic pigs exhibiting a Near Eastern genetic signature had all but disappeared across Anatolia and the southern Caucasus. Though we did not detect European signatures in the ancient Iranian samples (fig. 1), the eastward spread of European lineages may have continued into Iran later than the Iron Age since European lineages have been found in wild caught modern Iranian samples (Larson et al. 2007a).Of course a 12th century BC increase in European domestic pigs is entirely consistent -chronologically- with the Phrygian/Armenian settlement in Anatolia, and this association is further reinforced by the lack of European signatures in pigs from Iran where Phrygo-Armenians did not settle. The increase in European pigs could later be mediated by the Greek colonization, and the increase in trade during antiquity, just as trade would later introduce East Asian pig DNA into Europe.
The beautiful temporal transect presented in the Figure may also prove useful for students of ancient human DNA. I'd love to see how humans living close to sites #14-16, dominated by Arm1T haplotypes throughout history might differ from those of Neolithic West Anatolia, and whether the "mixed" Iron Age sample from Lidar Höyük shows evidence of the arrival of European-like human populations to accompany the European pigs.
Mol Biol Evol (2012) doi: 10.1093/molbev/mss261
Pig domestication and human-mediated dispersal in western Eurasia revealed through ancient DNA and geometric morphometrics
Claudio Ottoni et al.
Zooarcheological evidence suggests that pigs were domesticated in Southwest Asia ∼8,500 BC. They then spread across the Middle and Near East and westward into Europe alongside early agriculturalists. European pigs were either domesticated independently or appeared so as a result of admixture between introduced pigs and European wild boar. These pigs not only replaced those with Near Eastern signatures in Europe, they subsequently also replaced indigenous domestic pigs in the Near East. The specific details of these processes, however, remain unknown. To address questions related to early pig domestication, dispersal, and turnover in the Near East, we analyzed ancient mitochondrial DNA and dental geometric morphometric variation in 393 ancient pig specimens representing 48 archeological sites (from the Pre-Pottery Neolithic to the Medieval period) from Armenia, Cyprus, Georgia, Iran, Syria and Turkey. Our results firstly reveal the genetic signature of early domestic pigs in Eastern Turkey. We also demonstrate that these early pigs differed genetically from those in western Anatolia that were introduced to Europe during the Neolithic expansion. In addition, we present a significantly more refined chronology for the introduction of European domestic pigs into Asia Minor that took place during the Bronze Age, nearly 1,000 years earlier than previously detected. By the 5th century AD, European signatures completely replaced the endemic lineages possibly coinciding with the demographic and societal changes during the Anatolian Bronze and Iron Ages.
Link
November 22, 2012
ALDER signal of admixture in Ashkenazi Jews
(You can skip the first part if you want, and head straight to the RESULTS section)
Previous studies on uniparental markers have indicated that Ashkenazi Jews (AJ) were formed by admixture between a Near Eastern population and European host populations; the evidence for the former element seems pretty clear on the basis of Y-chromosomes where Jews possess a relatively high frequency of Y-haplogroup J1 (and a few others) that are quite rare in non-Jewish north/east Europeans. As for the latter, it seems probable on the basis of the location of Ashkenazi Jews on PCA plots where they tend to occupy an intermediate position between extant populations of the Levant (including Near Eastern Jews) and non-Jewish Europeans.
Anyone who has played around with genetic data will know that while AJ may be positioned in the aforementioned "intermediate" location within the "West Eurasian continuum" between Europe and Near East, they tend to form their own cluster at higher dimensions. And, indeed, this is why it's fairly easy for a clustering algorithm, such as my "Clusters Galore" (MCLUST/MDS) approach to pick out a very specific AJ cluster (e.g., here, or here, using a fastIBD approach). An Ashkenazi Jewish-specific cluster also pops out at higher K in ADMIXTURE analyses. This cluster may reflect endogamy within the AJ community until quite recent times.
One way of detecting admixture in a group is through the use of f3-statistics. The statistic f3(AJ; European, Near_East) could be negative --which would indicate admixture-- but it is usually not -at least in the combinations of (European, Near_East) I've tried, and this is consistent with either the presence admixture or absence of admixture.
A simple and intuitive way to see why post-admixture drift might mask the presence of admixture can be seen by means of a simple calculation. Remember that the f3-statistic's +/- sign depends on the +/- sign of quantities (c-a)*(c-b) where c is an allele frequency in the admixed (?) population we are investigating, and a, b in the two reference populations. We can pick a to be less than b with no loss of generality.
In the absence of strong drift (e.g., if all populations have a very large number of individuals), then the allele frequency c=xa+(1-x)b where x is the amount of admixture --between 0 and 1-- from group A and (1-x) from group B, and this c will be maintained little changed in the post-admixture phase. With the aid of a little algebra, we get that:
(c-a)*(c-b) = (xa+(1-x)b-a)*(xa+(1-x)b-b)
= (xa+b-xb-a)*(xa+b-xb-b) =
= x(x-1)(a-b)^2
and this is of course negative because we assumed that x was less than 1.
In a large population, this c will remain near-constant, because of the lack of strong drift. As long as it remains within the interval (a,b), then (c-a)*(c-b) will also remain negative, and so will the f3 statistic.
But, what if strong drift affects the admixed population? Allele frequencies fluctuate more wildly in larger populations, so c might go outside the (a,b) interval. Without loss of generality, assume that c becomes greater than b in which case (c-a)*(c-b) will become positive.
The f3-statistic averages over many SNPs, so, depending on (i) the initial differentiation of the admixed populations, which could be seen as b-a, and (ii) the amount of drift, which causes c to jump outside the (a, b) interval as discussed above, it is possible that the evidence for admixture may disappear.
So, relying on allele frequency differences may help obliterate the signal of admixture. But, there is a different signal of admixture that uses the decay of admixture linkage-disequilibrium, most recently discussed in the ALDER paper. The admixture LD signal's evidence may also disappear in time, but only because the signal occurs at increasingly lower genetic distances over time due to recombination. Thankfully, it tends to occur at large enough --for the last few thousand years-- distances, for which the SNP density of existing genotyping platforms that measure a few hundred thousand SNPs per individual is sufficient.
METHODS
Naturally I was curious to see whether the admixture LD mechanism would produce the evidence of admixture that the f3-statistics did not. I combined three datasets in my possession (HGDP by Li et al. Behar et al. and Yunusbayev et al. ) and identified sets of European and Semitic populations. (Remember that these sets are non-exhaustive, but presumably usable surrogates for the true mixing populations exist within them):
Abhkasians_Y, Adygei, Belorussian, Bulgarians_Y, Chechens_Y, Chuvashs, French, French_Basque, Georgians, Hungarians, Lezgins, Lithuanians, Mordovians_Y, North_Italian, North_Ossetians_Y, Orcadian, Romanians, Russian, Sardinian, Spaniards, Tuscan, Ukranians_Y
and:
Bedouin, Druze, Egyptans, Ethiopian_Jews, Ethiopians, Iraq_Jews, Jordanians, Lebanese, Morocco_Jews, Palestinian, Saudis, Sephardic_Jews, Syrians, Yemenese, Yemen_Jews
I used my Dodecad Project sample of AJ which numbers 36 individuals and is larger than any other usable public sample available to me.
(ALDER was run with default parameters, using the Rutgets recombination map for Illumina chips, and with the merged dataset prepared with a --geno 0.03 flag. Note that the Ashkenazi_D sample consists of individuals typed on different Illumina platforms from 23andMe and FamilyTreeDNA. The total number of SNPs considered was 527,165.)
RESULTS
I report below the tests for which ALDER reported "success" for the test with no warnings:

The median of all these estimates is 36.78 generations or 1070 years which corresponds to a calendar date of 910CE, assuming the sample's birthday was 1980, and a generation length of 29 years.
Palamara et al. placed the beginning of demographic expansion of AJ in a similar timeframe (33 generations), following a severe founder effect reducing the population to ~270 individuals. Such a founder effect may have indeed served to produce positive f3-statistics, masking the presence of admixture, the occurrence of which appears to be substantiated on the basis of the ALDER test of admixture.
As for the levels of admixture, using a 1-ref analysis with the European populations, I get the following lower bounds:

I'd be interested in hearing people's opinions on the plausibility of these dates/proportions, as well as their potential historical associations; a lot of factors might affect these results, so perhaps this analysis could be improved in the future.
Previous studies on uniparental markers have indicated that Ashkenazi Jews (AJ) were formed by admixture between a Near Eastern population and European host populations; the evidence for the former element seems pretty clear on the basis of Y-chromosomes where Jews possess a relatively high frequency of Y-haplogroup J1 (and a few others) that are quite rare in non-Jewish north/east Europeans. As for the latter, it seems probable on the basis of the location of Ashkenazi Jews on PCA plots where they tend to occupy an intermediate position between extant populations of the Levant (including Near Eastern Jews) and non-Jewish Europeans.
Anyone who has played around with genetic data will know that while AJ may be positioned in the aforementioned "intermediate" location within the "West Eurasian continuum" between Europe and Near East, they tend to form their own cluster at higher dimensions. And, indeed, this is why it's fairly easy for a clustering algorithm, such as my "Clusters Galore" (MCLUST/MDS) approach to pick out a very specific AJ cluster (e.g., here, or here, using a fastIBD approach). An Ashkenazi Jewish-specific cluster also pops out at higher K in ADMIXTURE analyses. This cluster may reflect endogamy within the AJ community until quite recent times.
One way of detecting admixture in a group is through the use of f3-statistics. The statistic f3(AJ; European, Near_East) could be negative --which would indicate admixture-- but it is usually not -at least in the combinations of (European, Near_East) I've tried, and this is consistent with either the presence admixture or absence of admixture.
A simple and intuitive way to see why post-admixture drift might mask the presence of admixture can be seen by means of a simple calculation. Remember that the f3-statistic's +/- sign depends on the +/- sign of quantities (c-a)*(c-b) where c is an allele frequency in the admixed (?) population we are investigating, and a, b in the two reference populations. We can pick a to be less than b with no loss of generality.
In the absence of strong drift (e.g., if all populations have a very large number of individuals), then the allele frequency c=xa+(1-x)b where x is the amount of admixture --between 0 and 1-- from group A and (1-x) from group B, and this c will be maintained little changed in the post-admixture phase. With the aid of a little algebra, we get that:
(c-a)*(c-b) = (xa+(1-x)b-a)*(xa+(1-x)b-b)
= (xa+b-xb-a)*(xa+b-xb-b) =
= x(x-1)(a-b)^2
and this is of course negative because we assumed that x was less than 1.
In a large population, this c will remain near-constant, because of the lack of strong drift. As long as it remains within the interval (a,b), then (c-a)*(c-b) will also remain negative, and so will the f3 statistic.
But, what if strong drift affects the admixed population? Allele frequencies fluctuate more wildly in larger populations, so c might go outside the (a,b) interval. Without loss of generality, assume that c becomes greater than b in which case (c-a)*(c-b) will become positive.
The f3-statistic averages over many SNPs, so, depending on (i) the initial differentiation of the admixed populations, which could be seen as b-a, and (ii) the amount of drift, which causes c to jump outside the (a, b) interval as discussed above, it is possible that the evidence for admixture may disappear.
So, relying on allele frequency differences may help obliterate the signal of admixture. But, there is a different signal of admixture that uses the decay of admixture linkage-disequilibrium, most recently discussed in the ALDER paper. The admixture LD signal's evidence may also disappear in time, but only because the signal occurs at increasingly lower genetic distances over time due to recombination. Thankfully, it tends to occur at large enough --for the last few thousand years-- distances, for which the SNP density of existing genotyping platforms that measure a few hundred thousand SNPs per individual is sufficient.
METHODS
Naturally I was curious to see whether the admixture LD mechanism would produce the evidence of admixture that the f3-statistics did not. I combined three datasets in my possession (HGDP by Li et al. Behar et al. and Yunusbayev et al. ) and identified sets of European and Semitic populations. (Remember that these sets are non-exhaustive, but presumably usable surrogates for the true mixing populations exist within them):
Abhkasians_Y, Adygei, Belorussian, Bulgarians_Y, Chechens_Y, Chuvashs, French, French_Basque, Georgians, Hungarians, Lezgins, Lithuanians, Mordovians_Y, North_Italian, North_Ossetians_Y, Orcadian, Romanians, Russian, Sardinian, Spaniards, Tuscan, Ukranians_Y
and:
Bedouin, Druze, Egyptans, Ethiopian_Jews, Ethiopians, Iraq_Jews, Jordanians, Lebanese, Morocco_Jews, Palestinian, Saudis, Sephardic_Jews, Syrians, Yemenese, Yemen_Jews
I used my Dodecad Project sample of AJ which numbers 36 individuals and is larger than any other usable public sample available to me.
(ALDER was run with default parameters, using the Rutgets recombination map for Illumina chips, and with the merged dataset prepared with a --geno 0.03 flag. Note that the Ashkenazi_D sample consists of individuals typed on different Illumina platforms from 23andMe and FamilyTreeDNA. The total number of SNPs considered was 527,165.)
RESULTS
I report below the tests for which ALDER reported "success" for the test with no warnings:
The median of all these estimates is 36.78 generations or 1070 years which corresponds to a calendar date of 910CE, assuming the sample's birthday was 1980, and a generation length of 29 years.
Palamara et al. placed the beginning of demographic expansion of AJ in a similar timeframe (33 generations), following a severe founder effect reducing the population to ~270 individuals. Such a founder effect may have indeed served to produce positive f3-statistics, masking the presence of admixture, the occurrence of which appears to be substantiated on the basis of the ALDER test of admixture.
As for the levels of admixture, using a 1-ref analysis with the European populations, I get the following lower bounds:
I'd be interested in hearing people's opinions on the plausibility of these dates/proportions, as well as their potential historical associations; a lot of factors might affect these results, so perhaps this analysis could be improved in the future.
November 20, 2012
Who inhabited the Jubbah lake in the Nefud Desert during the Middle Paleolithic?
Many readers may have heard of the Nefud Desert while watching Lawrence of Arabia (was that filmed on location?). It is hard to imagine that desolate landscape as being instrumental in the tale of human origins, but it may very well have been. A new paper describes Middle Paleolithic settlement evidence from the Jubbah Palaeolake, especially during MIS stages 7 and 5.
Arabia is a very interesting case for a variety of reasons: It has to be implicated one way or another in the tale of human origins and dispersals: it lies in the natural route Out-of-Africa, and in the intermediate space between the early modern human remains from Ethiopia, the later modern humans from the Levant, as well as the disputed late Neandertals of West Asia.
Unfortunately, current climatic conditions, as well as past episodes desiccation have resulted in substantial population; if anyone wanted to find out what the people who lived there during the Middle Paleolithic were like, he will find little continuity between them and the current inhabitants. The lack of genetic evidence is, unfortunately also accompanied by a general lack of anthropological evidence. Industries with links to Africa or the Levant are devoid of associated remains. But, the paper produces a hopeful note:
 In the absence of genes or bones, we can only make inferences based on stones, which may not have a direct correspondence with populations. While Figure 17 from the paper (left) shows a clear differentiation of India vis a vis. the west, relationships in the Near East and Africa are not as clear cut; Skhul resembles North Africa (Haua Fteah and Aterian) and it would be tempting to associate them with Homo sapiens. But, Horn of Africa MSA -where the earliest anatomically modern humans were found- is linked to El Wad, Tabun C, and Jebel Qattar/Katefeh, the latter two sites being the ones from the Nefud.
In the absence of genes or bones, we can only make inferences based on stones, which may not have a direct correspondence with populations. While Figure 17 from the paper (left) shows a clear differentiation of India vis a vis. the west, relationships in the Near East and Africa are not as clear cut; Skhul resembles North Africa (Haua Fteah and Aterian) and it would be tempting to associate them with Homo sapiens. But, Horn of Africa MSA -where the earliest anatomically modern humans were found- is linked to El Wad, Tabun C, and Jebel Qattar/Katefeh, the latter two sites being the ones from the Nefud.
Tabun is associated with Neandertals, although that attribution, like most everything in palaeoanthropology is controversial.So, it might be possible that the Jubbah was occupied by Neandertals too, and this might make this population a prime candidate for the signal of Neandertal admixture carried by non-Africans.
At present, there seem to be two candidates for the modern human Out-of-Africa: Skhul (Levant; linked to Northwest Africa here) and the Nubian technocomplex of (south Arabia; linked to Northeast Africa). I don't have a clear picture of how it may have all played out; it would certainly be wonderful if it were possible to extract DNA from, say, Skhul/Qafzeh modern humans or the Levantine Neandertals, because that would definitely show how (i) the former may either be related to later Eurasians, or may be a failed experiment as hitherto supposed, and (ii) the latter might be a source of Neandertal DNA in non-Africans, or indeed something much closer to modern humans as their morphological intermediacy might suggest.
PLoS ONE 7(11): e49840. doi:10.1371/journal.pone.0049840
Hominin Dispersal into the Nefud Desert and Middle Palaeolithic Settlement along the Jubbah Palaeolake, Northern Arabia
Michael D. Petraglia et al.
The Arabian Peninsula is a key region for understanding hominin dispersals and the effect of climate change on prehistoric demography, although little information on these topics is presently available owing to the poor preservation of archaeological sites in this desert environment. Here, we describe the discovery of three stratified and buried archaeological sites in the Nefud Desert, which includes the oldest dated occupation for the region. The stone tool assemblages are identified as a Middle Palaeolithic industry that includes Levallois manufacturing methods and the production of tools on flakes. Hominin occupations correspond with humid periods, particularly Marine Isotope Stages 7 and 5 of the Late Pleistocene. The Middle Palaeolithic occupations were situated along the Jubbah palaeolake-shores, in a grassland setting with some trees. Populations procured different raw materials across the lake region to manufacture stone tools, using the implements to process plants and animals. To reach the Jubbah palaeolake, Middle Palaeolithic populations travelled into the ameliorated Nefud Desert interior, possibly gaining access from multiple directions, either using routes from the north and west (the Levant and the Sinai), the north (the Mesopotamian plains and the Euphrates basin), or the east (the Persian Gulf). The Jubbah stone tool assemblages have their own suite of technological characters, but have types reminiscent of both African Middle Stone Age and Levantine Middle Palaeolithic industries. Comparative inter-regional analysis of core technology indicates morphological similarities with the Levantine Tabun C assemblage, associated with human fossils controversially identified as either Neanderthals or Homo sapiens.
Link
Arabia is a very interesting case for a variety of reasons: It has to be implicated one way or another in the tale of human origins and dispersals: it lies in the natural route Out-of-Africa, and in the intermediate space between the early modern human remains from Ethiopia, the later modern humans from the Levant, as well as the disputed late Neandertals of West Asia.
Unfortunately, current climatic conditions, as well as past episodes desiccation have resulted in substantial population; if anyone wanted to find out what the people who lived there during the Middle Paleolithic were like, he will find little continuity between them and the current inhabitants. The lack of genetic evidence is, unfortunately also accompanied by a general lack of anthropological evidence. Industries with links to Africa or the Levant are devoid of associated remains. But, the paper produces a hopeful note:
Yet, recent support for an MIS 5 expansion of Homo sapiens comes from archaeological finds of characteristic Middle Palaeolithic technologies in Arabia in MIS 5e–c [19]–[20] and nuclear genomic estimates which indicate that the split between Africans and non-Africans occurred as early as 130 to 90 ka [41], consistent with fossil finds of Homo sapiens in the Levant [52], [53] and at the time of possible interbreeding of Homo sapiens and Neanderthals [54]. These controversies indicate the need to recover hominin fossils in Arabia, which is feasible given the identification of Pleistocene mammalian fauna in a nearby lake basin of the Nefud [24], [55].

Tabun is associated with Neandertals, although that attribution, like most everything in palaeoanthropology is controversial.So, it might be possible that the Jubbah was occupied by Neandertals too, and this might make this population a prime candidate for the signal of Neandertal admixture carried by non-Africans.
At present, there seem to be two candidates for the modern human Out-of-Africa: Skhul (Levant; linked to Northwest Africa here) and the Nubian technocomplex of (south Arabia; linked to Northeast Africa). I don't have a clear picture of how it may have all played out; it would certainly be wonderful if it were possible to extract DNA from, say, Skhul/Qafzeh modern humans or the Levantine Neandertals, because that would definitely show how (i) the former may either be related to later Eurasians, or may be a failed experiment as hitherto supposed, and (ii) the latter might be a source of Neandertal DNA in non-Africans, or indeed something much closer to modern humans as their morphological intermediacy might suggest.
PLoS ONE 7(11): e49840. doi:10.1371/journal.pone.0049840
Hominin Dispersal into the Nefud Desert and Middle Palaeolithic Settlement along the Jubbah Palaeolake, Northern Arabia
Michael D. Petraglia et al.
The Arabian Peninsula is a key region for understanding hominin dispersals and the effect of climate change on prehistoric demography, although little information on these topics is presently available owing to the poor preservation of archaeological sites in this desert environment. Here, we describe the discovery of three stratified and buried archaeological sites in the Nefud Desert, which includes the oldest dated occupation for the region. The stone tool assemblages are identified as a Middle Palaeolithic industry that includes Levallois manufacturing methods and the production of tools on flakes. Hominin occupations correspond with humid periods, particularly Marine Isotope Stages 7 and 5 of the Late Pleistocene. The Middle Palaeolithic occupations were situated along the Jubbah palaeolake-shores, in a grassland setting with some trees. Populations procured different raw materials across the lake region to manufacture stone tools, using the implements to process plants and animals. To reach the Jubbah palaeolake, Middle Palaeolithic populations travelled into the ameliorated Nefud Desert interior, possibly gaining access from multiple directions, either using routes from the north and west (the Levant and the Sinai), the north (the Mesopotamian plains and the Euphrates basin), or the east (the Persian Gulf). The Jubbah stone tool assemblages have their own suite of technological characters, but have types reminiscent of both African Middle Stone Age and Levantine Middle Palaeolithic industries. Comparative inter-regional analysis of core technology indicates morphological similarities with the Levantine Tabun C assemblage, associated with human fossils controversially identified as either Neanderthals or Homo sapiens.
Link
U7 in Rostov Scythians
I found it quite interesting that in terms of mtDNA, the Rostov Scythians studied by der Sarkissian resembled closely the Shugnans of Tajikistan, who speak an eastern Iranian language. The author finds links between the Scythians and the "Central Asian Corridor", in particular with respect to mtDNA haplogroup U7.
This "Central Asian Corridor" sensu der Sarkissian (Iraq, Iran, Pakistan, India) seems to touch Frachetti's Inner Asian Mountain Corridor (shown below) in the region of the Pamirs.


Interestingly, the Sughnans belong, anthropologically to the Pamir-Ferghana type, which was also called Central Asian interfluvial type, the rivers in question being the Oxus and Jaxartes (Amu Darya and Syr Darya). And, of course, between these two rivers was the heartland of the Bactria Margiana Archaeological Complex, which I have previously linked with the Indo-Iranians.
Wells et al. studied Y-chromosomes of Sughnans, Yagnobis and other Iranic survivals of Tajikistan more than 10 years ago, and it will be very well worth revisiting them with newer methods. The area east of the Caspian and west of the IAMC intersects so much history, that any data from from it (new or ancient) would be extremely useful.
In my own experiments there has been an unambiguous "South Asian" genetic component in almost all Iranic peoples, even the westernmost Kurds. While the interpretation of this component is not easy, it does point to a genetic relationship between its possessors and Central/South Asia, with notable contrasts between Kurds/Iranians and their non-Iranic Armenian/Anatolian/Caucasian neighbors.
The occurrence of mtDNA haplogroup U7 in the Rostov Scythians is also consistent with a link between the Iranian nomads who penetrated into Europe with the area east of the Caspian, and it is also, of course, consistent with the narrative of Herodotus who recorded the migration of the Scythians into Europe.
There is a widely held theory that the origin of the Indo-Iranians are to be sought in eastern Europe. That theory appears inconsistent both with the "South Asian" autosomal signal in Iranic groups, and with the mtDNA evidence. Consider, again, the evidence of der Sarkissian:


Now, if Rostov Scythians were primarily descended from Mesolithic West Eurasians or even Bronze Age ones, then we would expect them to cluster at the "top", approaching the northern Europeoid extrema of PWC and Bronze Age Altai (ALT-BA). On the contrary, their position is well to the "south" of all European Bronze Age groups, and intermediate between Europeans and Iron Age Asian groups from south Siberia and Kazakhstan (KUR-IA, KAZ-IA). Again, this is compatible with an east-west migration during the Iron Age.
It might be worth speculating on the possible autosomal history of the steppe, for which the mtDNA evidence complements others: I offer that the long-term trend will be one of diminishing "North European", increasing "West Asian" and "East Eurasian" influences across the Neolithic-Bronze-Iron Age boundaries. At the western end of the steppe, there may also be "Mediterranean"/Sardinian-like infusions from the Balkans and Central Europe, although these clearly did not influence Inner/South Asia (where Mediterranean components shrink to non-existence), and Europe proper was mostly the recipient rather than the emitter of populations to Asia. Hopefully, autosomal data to test this conjecture will be made available in the coming years.
This "Central Asian Corridor" sensu der Sarkissian (Iraq, Iran, Pakistan, India) seems to touch Frachetti's Inner Asian Mountain Corridor (shown below) in the region of the Pamirs.


Interestingly, the Sughnans belong, anthropologically to the Pamir-Ferghana type, which was also called Central Asian interfluvial type, the rivers in question being the Oxus and Jaxartes (Amu Darya and Syr Darya). And, of course, between these two rivers was the heartland of the Bactria Margiana Archaeological Complex, which I have previously linked with the Indo-Iranians.
Wells et al. studied Y-chromosomes of Sughnans, Yagnobis and other Iranic survivals of Tajikistan more than 10 years ago, and it will be very well worth revisiting them with newer methods. The area east of the Caspian and west of the IAMC intersects so much history, that any data from from it (new or ancient) would be extremely useful.
In my own experiments there has been an unambiguous "South Asian" genetic component in almost all Iranic peoples, even the westernmost Kurds. While the interpretation of this component is not easy, it does point to a genetic relationship between its possessors and Central/South Asia, with notable contrasts between Kurds/Iranians and their non-Iranic Armenian/Anatolian/Caucasian neighbors.
The occurrence of mtDNA haplogroup U7 in the Rostov Scythians is also consistent with a link between the Iranian nomads who penetrated into Europe with the area east of the Caspian, and it is also, of course, consistent with the narrative of Herodotus who recorded the migration of the Scythians into Europe.
There is a widely held theory that the origin of the Indo-Iranians are to be sought in eastern Europe. That theory appears inconsistent both with the "South Asian" autosomal signal in Iranic groups, and with the mtDNA evidence. Consider, again, the evidence of der Sarkissian:


Now, if Rostov Scythians were primarily descended from Mesolithic West Eurasians or even Bronze Age ones, then we would expect them to cluster at the "top", approaching the northern Europeoid extrema of PWC and Bronze Age Altai (ALT-BA). On the contrary, their position is well to the "south" of all European Bronze Age groups, and intermediate between Europeans and Iron Age Asian groups from south Siberia and Kazakhstan (KUR-IA, KAZ-IA). Again, this is compatible with an east-west migration during the Iron Age.
It might be worth speculating on the possible autosomal history of the steppe, for which the mtDNA evidence complements others: I offer that the long-term trend will be one of diminishing "North European", increasing "West Asian" and "East Eurasian" influences across the Neolithic-Bronze-Iron Age boundaries. At the western end of the steppe, there may also be "Mediterranean"/Sardinian-like infusions from the Balkans and Central Europe, although these clearly did not influence Inner/South Asia (where Mediterranean components shrink to non-existence), and Europe proper was mostly the recipient rather than the emitter of populations to Asia. Hopefully, autosomal data to test this conjecture will be made available in the coming years.
November 19, 2012
Mitochondrial DNA in Ancient Human Populations of Europe (der Sarkissian 2011)
Going over the 322 pages of thesis may take a while, but feel free to comment on it if you discover any interesting nuggets in the text. The following view of West/East Eurasian mtDNA surrounding the beginning of the Iron Age may be useful, and seems to parallel the results of a recent paper on Pazyryk mtDNA:

Of course, since the thesis was published we have new data from West Siberia/Ukraine that suggest that the penetration of east Eurasian lineages covered a great area to the west of the indicated region even prior to the Iron Age.
We can be fairly sure that "non-East Eurasian admixed" populations existed during the Bronze Age in three portions of the Eurasian landmass, separated by the Black and Caspian Seas: west of the Black Sea (Balkans/Central Europe); between Black and Caspian Seas (Caucasus) and east of the Caspian Sea (Kazakhstan and Turkmenistan). But how did these three regions contribute to the West Eurasian elements found on a west-east axis across Eurasia today? And, to what extent did the early east Eurasian elements that penetrated well into eastern Europe in the Neolithic-to-Bronze Age contribute to latter populations of the area vs. more recent expansions from the Altai and Central Asia during the Iron Age?
Here is a PCA of the pre-Iron Age individuals, compared with modern populations:
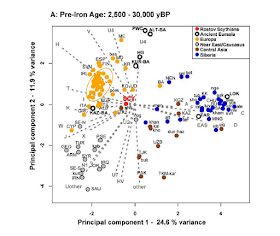
Both "Tarim" (TAR) and "Neolithic Lake Baikal" (LOK) appear well within east Eurasian variation. But, of the West Eurasian groups, Pitted Ware Complex (PWC), i.e., Neolithic hunter-gatherers from NE Europe and Bronze Age Altai (ALT-BA) appear clearly "northern Europeoid" across the 2nd PC, as do, to a lesser extent, C/N European Hunter-Gatherers (HG) and Kurgan burials from south Siberia (KUR-BA), but Bronze Age Kazakhstan (KAZ-BA) appear to be southern Europeoid, and, also, noticeably more "West Eurasian" than the others. Clearly, the West Eurasian elements were not homogeneous, with some of them (such as KAZ-BA) apparently derived from the southern Caucasoid zone -which largely did not experience east Eurasian admixture- and others from the northern Caucasoid zone that did.
The Rostov Scythian sample (in red) appears to belong to the southern Caucasoid zone (across PC2), but East Eurasian-shifted relative to modern Europeans and Bronze Age Kazakhstan.
Now, let's look at the Iron and post-Iron Age samples:

Egyin Gol (EG) from Mongolia and Sargat Siberians appear clearly as East Eurasians; Pazyryk Altai (ALT-IA), Iron Age Kazakhstan (KAZ-IA) and South Siberia Kurgan (KUR-IA) show decreasing East Eurasian influence; also notice the decidedly "southern" shift of the West Eurasian element among them.
This seems broadly consistent with the ideas of Molodin et al. about the gradual appearance (in their Siberian sample) of Caucasoid mtDNA types from the Neolithic to the Iron Age, with the early Neolithic U-dominated population finally receiving a full set of diverse West Eurasian lineages only during the Iron Age from the south.
It will certainly be very exciting when samples such as these can be tested for autosomal or Y-chromosome DNA, and I'm looking forward to the day when this can be done on a large scale.
Type: Thesis
Title: Mitochondrial DNA in ancient human populations of Europe.
Author: Dersarkissian, Clio Simone Irmgard
Issue Date: 2011
School/Discipline: School of Earth and Environmental Sciences
Abstract: The distribution of human genetic variability is the result of thousand years of human evolutionary and population history. Geographical variation in the nonrecombining maternally inherited mitochondrial DNA has been studied in a wide array of modern populations in order to reconstruct the migrations that have participated in the spread of our ancestors on the planet. However, population genetic processes (e.g., replacement, genetic drift) can significantly bias the reconstruction and timing of past migratory and demographic events inferred from the analysis of modern-day marker distributions. This can lead to erroneous interpretations of ancient human population history, a problem that potentially could be circumvented by the direct assessment of genetic diversity in ancient humans. Despite important methodological problems associated with contamination and post-mortem degradation of ancient DNA, mitochondrial data have been previously obtained for a few spatially and temporally diverse European populations. Mitochondrial data revealed additional levels of complexity in the population history of Europeans that had remained unknown from the study of modern populations. This justifies the relevance of broadening the sampling of ancient mitochondrial DNA in both time and space. This study aims at filling gaps in the knowledge of the genetic history of eastern Europeans and of European genetic outliers, the Saami and the Sardinians. This study presents a significant extension to the knowledge of past human mitochondrial diversity. Ancient remains temporally-sampled from three groups of European populations have been examined: north east Europeans (200 – 8,000 years before present; N = 76), Iron Age Scythians of the Rostov area, Russia (2,300 – 2,600 years before present; N = 16), Bronze Age individuals of central Sardinia, Italy (3,200 – 3,400 years before present; N = 16). The genetic characterisation of these populations principally relied on sequencing of the mitochondrial control region and typing of single nucleotide polymorphisms in the coding region. Changes in mitochondrial DNA structure were tracked through time by comparing ancient and modern populations of Eurasia. Analysis of haplogroup data included principal component analysis, multidimensional scaling, fixation index computation and genetic distance mapping. Haplotypic data were compared by haplotype sharing analysis, phylogenetic networks, Analysis of the Molecular Variance and coalescent simulations. The sequencing of a whole mitochondrial genome in a north east European Mesolithic individual lead to defining a new branch within the human mitochondrial tree. This work presents direct evidence that Mesolithic eastern Europeans belonged to the same Palaeolithic/Mesolithic genetic background as central and northern Europeans. It was also shown that prehistoric eastern Europeans were the recipients of multiple migrations from the East in prehistory that had not been previously detected and/or timed on the basis of modern mtDNA data. Ancient DNA also provided insights in the genetic history of European genetic outliers; the Saami, whose ancestral population still remain unidentified, and the Sardinians, whose genetic differentiation is proposed to be the result of mating isolation since at least the Bronze Age. This study demonstrates the power of aDNA to reveal previously unknown population processes in the genetic history of modern Eurasians.
Link

Of course, since the thesis was published we have new data from West Siberia/Ukraine that suggest that the penetration of east Eurasian lineages covered a great area to the west of the indicated region even prior to the Iron Age.
We can be fairly sure that "non-East Eurasian admixed" populations existed during the Bronze Age in three portions of the Eurasian landmass, separated by the Black and Caspian Seas: west of the Black Sea (Balkans/Central Europe); between Black and Caspian Seas (Caucasus) and east of the Caspian Sea (Kazakhstan and Turkmenistan). But how did these three regions contribute to the West Eurasian elements found on a west-east axis across Eurasia today? And, to what extent did the early east Eurasian elements that penetrated well into eastern Europe in the Neolithic-to-Bronze Age contribute to latter populations of the area vs. more recent expansions from the Altai and Central Asia during the Iron Age?
Here is a PCA of the pre-Iron Age individuals, compared with modern populations:

Both "Tarim" (TAR) and "Neolithic Lake Baikal" (LOK) appear well within east Eurasian variation. But, of the West Eurasian groups, Pitted Ware Complex (PWC), i.e., Neolithic hunter-gatherers from NE Europe and Bronze Age Altai (ALT-BA) appear clearly "northern Europeoid" across the 2nd PC, as do, to a lesser extent, C/N European Hunter-Gatherers (HG) and Kurgan burials from south Siberia (KUR-BA), but Bronze Age Kazakhstan (KAZ-BA) appear to be southern Europeoid, and, also, noticeably more "West Eurasian" than the others. Clearly, the West Eurasian elements were not homogeneous, with some of them (such as KAZ-BA) apparently derived from the southern Caucasoid zone -which largely did not experience east Eurasian admixture- and others from the northern Caucasoid zone that did.
The Rostov Scythian sample (in red) appears to belong to the southern Caucasoid zone (across PC2), but East Eurasian-shifted relative to modern Europeans and Bronze Age Kazakhstan.
Now, let's look at the Iron and post-Iron Age samples:

Egyin Gol (EG) from Mongolia and Sargat Siberians appear clearly as East Eurasians; Pazyryk Altai (ALT-IA), Iron Age Kazakhstan (KAZ-IA) and South Siberia Kurgan (KUR-IA) show decreasing East Eurasian influence; also notice the decidedly "southern" shift of the West Eurasian element among them.
This seems broadly consistent with the ideas of Molodin et al. about the gradual appearance (in their Siberian sample) of Caucasoid mtDNA types from the Neolithic to the Iron Age, with the early Neolithic U-dominated population finally receiving a full set of diverse West Eurasian lineages only during the Iron Age from the south.
It will certainly be very exciting when samples such as these can be tested for autosomal or Y-chromosome DNA, and I'm looking forward to the day when this can be done on a large scale.
Type: Thesis
Title: Mitochondrial DNA in ancient human populations of Europe.
Author: Dersarkissian, Clio Simone Irmgard
Issue Date: 2011
School/Discipline: School of Earth and Environmental Sciences
Abstract: The distribution of human genetic variability is the result of thousand years of human evolutionary and population history. Geographical variation in the nonrecombining maternally inherited mitochondrial DNA has been studied in a wide array of modern populations in order to reconstruct the migrations that have participated in the spread of our ancestors on the planet. However, population genetic processes (e.g., replacement, genetic drift) can significantly bias the reconstruction and timing of past migratory and demographic events inferred from the analysis of modern-day marker distributions. This can lead to erroneous interpretations of ancient human population history, a problem that potentially could be circumvented by the direct assessment of genetic diversity in ancient humans. Despite important methodological problems associated with contamination and post-mortem degradation of ancient DNA, mitochondrial data have been previously obtained for a few spatially and temporally diverse European populations. Mitochondrial data revealed additional levels of complexity in the population history of Europeans that had remained unknown from the study of modern populations. This justifies the relevance of broadening the sampling of ancient mitochondrial DNA in both time and space. This study aims at filling gaps in the knowledge of the genetic history of eastern Europeans and of European genetic outliers, the Saami and the Sardinians. This study presents a significant extension to the knowledge of past human mitochondrial diversity. Ancient remains temporally-sampled from three groups of European populations have been examined: north east Europeans (200 – 8,000 years before present; N = 76), Iron Age Scythians of the Rostov area, Russia (2,300 – 2,600 years before present; N = 16), Bronze Age individuals of central Sardinia, Italy (3,200 – 3,400 years before present; N = 16). The genetic characterisation of these populations principally relied on sequencing of the mitochondrial control region and typing of single nucleotide polymorphisms in the coding region. Changes in mitochondrial DNA structure were tracked through time by comparing ancient and modern populations of Eurasia. Analysis of haplogroup data included principal component analysis, multidimensional scaling, fixation index computation and genetic distance mapping. Haplotypic data were compared by haplotype sharing analysis, phylogenetic networks, Analysis of the Molecular Variance and coalescent simulations. The sequencing of a whole mitochondrial genome in a north east European Mesolithic individual lead to defining a new branch within the human mitochondrial tree. This work presents direct evidence that Mesolithic eastern Europeans belonged to the same Palaeolithic/Mesolithic genetic background as central and northern Europeans. It was also shown that prehistoric eastern Europeans were the recipients of multiple migrations from the East in prehistory that had not been previously detected and/or timed on the basis of modern mtDNA data. Ancient DNA also provided insights in the genetic history of European genetic outliers; the Saami, whose ancestral population still remain unidentified, and the Sardinians, whose genetic differentiation is proposed to be the result of mating isolation since at least the Bronze Age. This study demonstrates the power of aDNA to reveal previously unknown population processes in the genetic history of modern Eurasians.
Link
November 17, 2012
Populations histories with a diffusion process formulation

Mol Biol Evol (2012) doi: 10.1093/molbev/mss257
Inferring population histories using genome-wide allele frequency data
Mathieu Gautier and Renaud Vitalis
The recent development of high throughput genotyping technologies has revolutionized the collection of data in a wide range of both model and non-model species. These data generally contain huge amounts of information about the past demographic history of populations.
In this study we introduce a new method to estimate divergence times on a diffusion time-scale from large SNP datasets, conditionally on a population history which is represented as a tree. We further assume that all the observed polymorphisms originate from the most ancestral (root) population, i.e. we neglect mutations that occur after the split of the most ancestral population. This method relies on a hierarchical-Bayesian model, based on Kimura's time-dependent diffusion approximation of genetic drift. We implemented a Metropolis–Hastings within Gibbs sampler to estimate the posterior distribution of the parameters of interest in this model, which we refer to as the Kimura model. Evaluating the Kimura model on simulated population histories, we found that it provides accurate estimates of divergence time. Assessing model fit using the deviance information criterion (DIC) proved efficient for retrieving the correct tree topology among a set of competing histories. We show that this procedure is robust to low-to-moderate gene flow, as well as to ascertainment bias, providing that the most distantly related populations are represented in the discovery panel. As an illustrative example, we finally analyzed published human data consisting in genotypes for 452,198 SNPs from individuals belonging to four populations worldwide.
Our results suggest that the Kimura model may be helpful to characterize the demographic history of dierentiated populations, using genome-wide allele frequency data.
Link
November 16, 2012
f3-statistics on craniometric data?
It occurred to me that the concept of f3-statistics, originally developed to detect admixture by exploiting allele frequency difference anti-correlations could very well be applied to craniometric data as well.
The basic idea is quite simple: suppose that for a metric trait, two populations A and B have mean value a and b and that a third population C is formed by mixture between A and B. Unlike allele frequencies where the admixed population's frequency will be between a and b immediately post-admixture, anthropometric traits may respond in unexpected ways to admixture (e.g., heterosis might cause first-generation offspring to exceed both their parents in height, rather than exhibit an intermediate value). I will leave the justification of the hypothesis that "mixed-origin offspring will possess intermediate metric traits" to the physical anthropologists, who may have gathered data on such things, and, for the present, I will take it for granted.
So, assuming that c, the mean trait in the mixed population, is between a and b, we can easily see that (c-a)(c-b) will be negative, and hence so will be the correlation coefficient (over many traits) between C-A and C-B, where by C-A I denote the k-long vector difference of mean trait values between populations C and A.
Going back to my analysis of Howells' dataset, I calculated population means for 57 traits over the NORMALIZED_DATA array of modern populations (in which sexual dimorphism has been removed and traits of different scale have been normalized in standard deviation units), and calculated 30*choose(29,2) correlations for each of 30 populations, expressed as a mixture of any pair of the remaining 29.
I list below, the top 20 anti-correlations, and highlight a few in bold (third population as mixture of first two):
BURIAT ANDAMAN PHILLIPI -0.54005191575771
EGYPT BURIAT NORSE -0.490018084440697
ANDAMAN ANYANG HAINAN -0.48323680182295
BURIAT ANDAMAN HAINAN -0.480939028739347
EGYPT BURIAT ZALAVAR -0.476445836100052
ANDAMAN ANYANG PHILLIPI -0.457902384166767
DOGON BURIAT PHILLIPI -0.416551851781419
BERG EASTER_I ZALAVAR -0.378996437433417
AUSTRALI BURIAT ARIKARA -0.375898166338775
BURIAT EASTER_I MOKAPU -0.37169703838378
ESKIMO ANDAMAN S_JAPAN -0.366611599944932
ESKIMO PERU N_JAPAN -0.354535077363928
TOLAI BURIAT ARIKARA -0.348110323746154
BERG EGYPT ZALAVAR -0.344843098962355
DOGON ESKIMO GUAM -0.344577928128792
TOLAI BURIAT GUAM -0.338804214799388
ESKIMO PHILLIPI GUAM -0.336537918547276
DOGON BURIAT HAINAN -0.332635954428392
TASMANIA BURIAT ARIKARA -0.331301837598433
ESKIMO PERU S_JAPAN -0.330302035072489
The basic idea is quite simple: suppose that for a metric trait, two populations A and B have mean value a and b and that a third population C is formed by mixture between A and B. Unlike allele frequencies where the admixed population's frequency will be between a and b immediately post-admixture, anthropometric traits may respond in unexpected ways to admixture (e.g., heterosis might cause first-generation offspring to exceed both their parents in height, rather than exhibit an intermediate value). I will leave the justification of the hypothesis that "mixed-origin offspring will possess intermediate metric traits" to the physical anthropologists, who may have gathered data on such things, and, for the present, I will take it for granted.
So, assuming that c, the mean trait in the mixed population, is between a and b, we can easily see that (c-a)(c-b) will be negative, and hence so will be the correlation coefficient (over many traits) between C-A and C-B, where by C-A I denote the k-long vector difference of mean trait values between populations C and A.
Going back to my analysis of Howells' dataset, I calculated population means for 57 traits over the NORMALIZED_DATA array of modern populations (in which sexual dimorphism has been removed and traits of different scale have been normalized in standard deviation units), and calculated 30*choose(29,2) correlations for each of 30 populations, expressed as a mixture of any pair of the remaining 29.
I list below, the top 20 anti-correlations, and highlight a few in bold (third population as mixture of first two):
BURIAT ANDAMAN PHILLIPI -0.54005191575771
EGYPT BURIAT NORSE -0.490018084440697
ANDAMAN ANYANG HAINAN -0.48323680182295
BURIAT ANDAMAN HAINAN -0.480939028739347
EGYPT BURIAT ZALAVAR -0.476445836100052
ANDAMAN ANYANG PHILLIPI -0.457902384166767
DOGON BURIAT PHILLIPI -0.416551851781419
BERG EASTER_I ZALAVAR -0.378996437433417
AUSTRALI BURIAT ARIKARA -0.375898166338775
BURIAT EASTER_I MOKAPU -0.37169703838378
ESKIMO ANDAMAN S_JAPAN -0.366611599944932
ESKIMO PERU N_JAPAN -0.354535077363928
TOLAI BURIAT ARIKARA -0.348110323746154
BERG EGYPT ZALAVAR -0.344843098962355
DOGON ESKIMO GUAM -0.344577928128792
TOLAI BURIAT GUAM -0.338804214799388
ESKIMO PHILLIPI GUAM -0.336537918547276
DOGON BURIAT HAINAN -0.332635954428392
TASMANIA BURIAT ARIKARA -0.331301837598433
ESKIMO PERU S_JAPAN -0.330302035072489
Some interesting ones:
- Philippines as Buriat+Andaman; this makes sense if Philippines is the result of admixture between an "East Asian" and a "Negrito" population
- Norse as Egypt+Buriat; the Howells "Egypt" sample is "Mediterranean" in the classical sense. Perhaps this involves the same "East Eurasian"-like signal of admixture detected by genetic methods? Similar signal also occurs for Zalavar (from Hungary)
- Hainan as Andaman+Anyang; south Chinese as Neolithic Chinese+"Negrito"-like old south Chinese?
- Arikara as Buriat+Australian; admixture between "Australoid" Paleo-Indians and "Mongoloid" ones? or between 1st wave Indians and later ones (sensu Reich et al. 2012)?
- Guam as Tolai+Buriat; admixture between "Papuan"-like and East Asian-like people in Polynesia?
And, there are some difficult-to-interpret cases (e.g., Philippines as Buriat+Dogon) which may point to limitations of the method; for example, the Dogon may act as a stand-in for the "equatorial"-like physique of the true "Andaman"-like mixing element. Presumably such limitations can be overcome by limiting the analysis to "selectively neutral" traits, rather than the whole suite of 57 Howells variables used here.
I certainly think that the idea ought to be investigated further: it might be redundant when genetic data are available, but may prove useful in the analysis of admixture when such data do not exist, e.g., in anthropological data of prehistoric specimens from hot climates where archaeogenetic evidence may never materialize.
Pre-Neolithic Mediterranean Island settlement
PhysOrg coverage of a Science perspective:
Science 16 November 2012: Vol. 338 no. 6109 pp. 895-897 DOI: 10.1126/science.1228880
Mediterranean Island Voyages
Alan Simmons
Some of the classical world's most innovative cultures developed on Mediterranean islands, but their earlier human use is poorly known. The islands, particularly those further from the mainland such as Crete and Cyprus, were thought to have been first colonized about 9000 years ago by late Neolithic agriculturalists with domesticated resources. Until about 20 years ago, claims of earlier, pre-Neolithic occupations on any of the islands did not stand up to critical scrutiny (1), but current investigations are challenging these perceptions. Discoveries on Cyprus, Crete, and some Ionian islands suggest seafaring abilities by pre-Neolithic peoples, perhaps extending back to Neanderthals or even earlier hominins. In Cyprus, Neolithic sites have been documented that are nearly as early as those on the mainland.
Link
Modern science has held that islands such as Cyprus and Crete were first inhabited by seafaring humans approximately 9,000 years ago by agriculturists from the late Neolithic period. Simmons writes that research over the past 20 years has cast doubt on that assumption however and suggests that it might be time to rewrite the history books. He cites evidence such as pieces of obsidian found in a cave in mainland Greece that were found to have come from Melos, an island in the Aegean Sea and were dated at 11,000 years ago as well as artifacts from recent digs on Cyprus that are believed to be from approximately 12,000 years ago. He adds that some researchers have also found evidence that something, or someone caused the extinction of pygmy hippos on Cyprus around the same time.
Simmons also suggests that the first inhabitants of many of the Mediterranean islands may not have been modern humans at all. Instead, he says evidence has been found that shows that they might have been Neanderthals, or Homo Erectus. Recent excavations on Crete have turned up artifacts that are thought to be 110,000 years old, for example, and a stone axe was found that is believed to have been made on the same island as far back as 170,000 years ago. Since modern humans are believed to have come into being roughly 100,000 to 200,000 years ago, the possibility exists that such artifacts were left behind by an early ancestor or cousin.
Science 16 November 2012: Vol. 338 no. 6109 pp. 895-897 DOI: 10.1126/science.1228880
Mediterranean Island Voyages
Alan Simmons
Some of the classical world's most innovative cultures developed on Mediterranean islands, but their earlier human use is poorly known. The islands, particularly those further from the mainland such as Crete and Cyprus, were thought to have been first colonized about 9000 years ago by late Neolithic agriculturalists with domesticated resources. Until about 20 years ago, claims of earlier, pre-Neolithic occupations on any of the islands did not stand up to critical scrutiny (1), but current investigations are challenging these perceptions. Discoveries on Cyprus, Crete, and some Ionian islands suggest seafaring abilities by pre-Neolithic peoples, perhaps extending back to Neanderthals or even earlier hominins. In Cyprus, Neolithic sites have been documented that are nearly as early as those on the mainland.
Link
Effect of genomic inversions on population genetic parameters
Genetics doi: 10.1534/genetics.112.145599
The Effect of Genomic Inversions on Estimation of Population Genetic Parameters from SNP Data
Nafisa-Katrin Seich al Basatena et al.
In recent years it has emerged that structural variants have a substantial impact on genomic variation. Inversion polymorphisms represent a significant class of structural variant, and despite the challenges in their detection, data on inversions in the human genome are increasing rapidly. Statistical methods for inferring parameters such as the recombination rate and selection coefficient have generally been developed without accounting for the presence of inversions. Here we exploit new software for simulating inversions in population genetic data, invertFREGENE, to assess the potential impact of inversions on such methods. Using data simulated by invertFREGENE, as well as real data from several sources, we test whether large inversions have a disruptive effect on widely applied population genetics methods for inferring recombination rates, for detecting selection, and for controlling for population structure in genome-wide association studies (GWAS). We find that recombination rates estimated by LDhat are biased downward at inversion loci relative to the true contemporary recombination rates at the loci but that recombination hotspots are not falsely inferred at inversion breakpoints as may have been expected. We find that the iHS method for detecting selection appears robust to the presence of inversions. Finally, we observe a strong bias on the genome-wide results of principle components analysis (PCA), used to control for population structure in GWAS, in the presence of even a single large inversion, confirming the necessity to thin SNPs by LD at large physical distances in order to obtain unbiased results.
Link
The Effect of Genomic Inversions on Estimation of Population Genetic Parameters from SNP Data
Nafisa-Katrin Seich al Basatena et al.
In recent years it has emerged that structural variants have a substantial impact on genomic variation. Inversion polymorphisms represent a significant class of structural variant, and despite the challenges in their detection, data on inversions in the human genome are increasing rapidly. Statistical methods for inferring parameters such as the recombination rate and selection coefficient have generally been developed without accounting for the presence of inversions. Here we exploit new software for simulating inversions in population genetic data, invertFREGENE, to assess the potential impact of inversions on such methods. Using data simulated by invertFREGENE, as well as real data from several sources, we test whether large inversions have a disruptive effect on widely applied population genetics methods for inferring recombination rates, for detecting selection, and for controlling for population structure in genome-wide association studies (GWAS). We find that recombination rates estimated by LDhat are biased downward at inversion loci relative to the true contemporary recombination rates at the loci but that recombination hotspots are not falsely inferred at inversion breakpoints as may have been expected. We find that the iHS method for detecting selection appears robust to the presence of inversions. Finally, we observe a strong bias on the genome-wide results of principle components analysis (PCA), used to control for population structure in GWAS, in the presence of even a single large inversion, confirming the necessity to thin SNPs by LD at large physical distances in order to obtain unbiased results.
Link
First Polynesian settlement: 2838±8 BP

High Precision U/Th Dating of First Polynesian Settlement
David Burley et al.
Previous studies document Nukuleka in the Kingdom of Tonga as a founder colony for first settlement of Polynesia by Lapita peoples. A limited number of radiocarbon dates are one line of evidence supporting this claim, but they cannot precisely establish when this event occurred, nor can they afford a detailed chronology for sequent occupation. High precision U/Th dates of Acropora coral files (abraders) from Nukuleka give unprecedented resolution, identifying the founder event by 2838±8 BP and documenting site development over the ensuing 250 years. The potential for dating error due to post depositional diagenetic alteration of ancient corals at Nukuleka also is addressed through sample preparation protocols and paired dates on spatially separated samples for individual specimens. Acropora coral files are widely distributed in Lapita sites across Oceania. U/Th dating of these artifacts provides unparalleled opportunities for greater precision and insight into the speed and timing of this final chapter in human settlement of the globe.
Link
TreeMix paper "officially" published
~8 months after the paper was pre-published in Nature Precedings, it is also "officially" published in PLoS Genetics. In the meantime, I count 18 uses of the label TreeMix in my blog, which includes both uses of the treemix software itself and its auxiliary threepop and fourpop programs; I also wrote a small script that converts ADMIXTURE output into TreeMix format, and generally had a lot of fun using it. I'm glad I didn't have to wait 8 months to learn that something like TreeMix existed.
In the grand scheme of things, an 8-month head start may not be much, but consider that perhaps someone else might either have a use for TreeMix or the desire to build on it, and if they decide to make their research available prior to official publication, then, perhaps an additional few months might be gained. And, if someone else still decides to follow up on them then...
There are many arguments for immediate publication of research results, but I think that the potential for speeding up scientific progress is one of the best ones.
In the old days, it was really necessary to impose a delay between the time when a scientist placed a final full stop to his paper and the time it appeared on another scientist's desk: publication involved significant expenses of paper, ink, and labor, so the frivolous or erroneous had to be weeded out; dissemination involved expensive transport by carriage or boat; storage involved a building, and bookshelves, and additional cost.
All these costs have shrunk to insignificance; imposing delays to research dissemination now accounts to little more than placing a sleep() call in the unending loop of scientific advancement. And, the one remaining argument for post-review publication ("weeding out the frivolous or erroneous") carries little weight: pre-review publication is a better guarantor of quality by exposing research to many more eyes and minds that may scrutinize it more carefully, having rid themselves of the idea that "if it's published it must be good".
PLoS Genet 8(11): e1002967. doi:10.1371/journal.pgen.1002967
Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data
Joseph K. Pickrell1, Jonathan K. Pritchard
Many aspects of the historical relationships between populations in a species are reflected in genetic data. Inferring these relationships from genetic data, however, remains a challenging task. In this paper, we present a statistical model for inferring the patterns of population splits and mixtures in multiple populations. In our model, the sampled populations in a species are related to their common ancestor through a graph of ancestral populations. Using genome-wide allele frequency data and a Gaussian approximation to genetic drift, we infer the structure of this graph. We applied this method to a set of 55 human populations and a set of 82 dog breeds and wild canids. In both species, we show that a simple bifurcating tree does not fully describe the data; in contrast, we infer many migration events. While some of the migration events that we find have been detected previously, many have not. For example, in the human data, we infer that Cambodians trace approximately 16% of their ancestry to a population ancestral to other extant East Asian populations. In the dog data, we infer that both the boxer and basenji trace a considerable fraction of their ancestry (9% and 25%, respectively) to wolves subsequent to domestication and that East Asian toy breeds (the Shih Tzu and the Pekingese) result from admixture between modern toy breeds and “ancient” Asian breeds. Software implementing the model described here, called TreeMix, is available at http://treemix.googlecode.com.
Link
In the grand scheme of things, an 8-month head start may not be much, but consider that perhaps someone else might either have a use for TreeMix or the desire to build on it, and if they decide to make their research available prior to official publication, then, perhaps an additional few months might be gained. And, if someone else still decides to follow up on them then...
There are many arguments for immediate publication of research results, but I think that the potential for speeding up scientific progress is one of the best ones.
In the old days, it was really necessary to impose a delay between the time when a scientist placed a final full stop to his paper and the time it appeared on another scientist's desk: publication involved significant expenses of paper, ink, and labor, so the frivolous or erroneous had to be weeded out; dissemination involved expensive transport by carriage or boat; storage involved a building, and bookshelves, and additional cost.
All these costs have shrunk to insignificance; imposing delays to research dissemination now accounts to little more than placing a sleep() call in the unending loop of scientific advancement. And, the one remaining argument for post-review publication ("weeding out the frivolous or erroneous") carries little weight: pre-review publication is a better guarantor of quality by exposing research to many more eyes and minds that may scrutinize it more carefully, having rid themselves of the idea that "if it's published it must be good".
PLoS Genet 8(11): e1002967. doi:10.1371/journal.pgen.1002967
Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data
Joseph K. Pickrell1, Jonathan K. Pritchard
Many aspects of the historical relationships between populations in a species are reflected in genetic data. Inferring these relationships from genetic data, however, remains a challenging task. In this paper, we present a statistical model for inferring the patterns of population splits and mixtures in multiple populations. In our model, the sampled populations in a species are related to their common ancestor through a graph of ancestral populations. Using genome-wide allele frequency data and a Gaussian approximation to genetic drift, we infer the structure of this graph. We applied this method to a set of 55 human populations and a set of 82 dog breeds and wild canids. In both species, we show that a simple bifurcating tree does not fully describe the data; in contrast, we infer many migration events. While some of the migration events that we find have been detected previously, many have not. For example, in the human data, we infer that Cambodians trace approximately 16% of their ancestry to a population ancestral to other extant East Asian populations. In the dog data, we infer that both the boxer and basenji trace a considerable fraction of their ancestry (9% and 25%, respectively) to wolves subsequent to domestication and that East Asian toy breeds (the Shih Tzu and the Pekingese) result from admixture between modern toy breeds and “ancient” Asian breeds. Software implementing the model described here, called TreeMix, is available at http://treemix.googlecode.com.
Link