
December 25, 2016
November 04, 2016

October 28, 2016


August 06, 2016
China's Great Flood and the rise of the Xia dynasty

Many cultures trace their origins to the hazy horizon where history meets legend. In China's case, that blurry line occurs sometime between 2200 B.C.E. and 2000 B.C.E., when a legendary hero named Yu tamed Yellow River flooding and earned a mandate to become the founding emperor of the Xia dynasty, the country's first. That’s the story according to texts written long after the fact, and many Chinese believe their civilization started with emperor Yu. But archaeologists have been unable to find convincing evidence for either the flood or the Xia dynasty itself.
...
The massive flood “provides us with a tantalizing hint that the Xia dynasty might really have existed," says David Cohen, an archaeologist and co-author at National Taiwan University in Taipei. The devastating flood could have inundated settlements even a thousand or more kilometers downstream, he says, and created chaos from which a new political order emerged. This sequence of events neatly fits the legend of Yu controlling the flooding by dredging channels to confine the Yellow River and its tributaries. This feat, the ancient texts say, allowed him to claim a mandate as the first emperor of the Xia dynasty.
The timing is curiously coincidental. Around 1900 B.C.E., Cohen says, Chinese society was transitioning from the Neolithic to the Bronze age. The date also correlates with what is called the Erlitou culture, which is known from palace buildings and bronze smelting workshops discovered near Zhengzhou, about 2500 kilometers downstream from Jishi Gorge. Many scholars have argued that Erlitou is a manifestation of the elusive Xia dynasty, but a link is not firmly established.
Science 05 Aug 2016: Vol. 353, Issue 6299, pp. 579-582 DOI: 10.1126/science.aaf0842
Outburst flood at 1920 BCE supports historicity of China’s Great Flood and the Xia dynasty
Qinglong Wu
China’s historiographical traditions tell of the successful control of a Great Flood leading to the establishment of the Xia dynasty and the beginning of civilization. However, the historicity of the flood and Xia remain controversial. Here, we reconstruct an earthquake-induced landslide dam outburst flood on the Yellow River about 1920 BCE that ranks as one of the largest freshwater floods of the Holocene and could account for the Great Flood. This would place the beginning of Xia at ~1900 BCE, several centuries later than traditionally thought. This date coincides with the major transition from the Neolithic to Bronze Age in the Yellow River valley and supports hypotheses that the primary state-level society of the Erlitou culture is an archaeological manifestation of the Xia dynasty.
Link
July 19, 2016
Educational achievement predicted by DNA
Predicting 9% of educational achievement from DNA is quite good. The authors used genotype arrays, so there's obvious room for growth in rare variation that is not covered by such arrays.
I wonder when the public and policymakers will get wind of the fact that educational achievement is highly heritable and can even be somewhat predicted with existing DNA technology.
Genetic egalitarianism is an edifice on which too much has been invested and I doubt that it will go down without a fight. It's of course a great idea to optimize learning for the students you've got. But, at the end of the day there's only so much you can do to foster achievement in a trait that is mostly genetically determined.
Molecular Psychiatry advance online publication 19 July 2016; doi: 10.1038/mp.2016.107
Predicting educational achievement from DNA
S Selzam et al.
A genome-wide polygenic score (GPS), derived from a 2013 genome-wide association study (N=127,000), explained 2% of the variance in total years of education (EduYears). In a follow-up study (N=329,000), a new EduYears GPS explains up to 4%. Here, we tested the association between this latest EduYears GPS and educational achievement scores at ages 7, 12 and 16 in an independent sample of 5825 UK individuals. We found that EduYears GPS explained greater amounts of variance in educational achievement over time, up to 9% at age 16, accounting for 15% of the heritable variance. This is the strongest GPS prediction to date for quantitative behavioral traits. Individuals in the highest and lowest GPS septiles differed by a whole school grade at age 16. Furthermore, EduYears GPS was associated with general cognitive ability (~3.5%) and family socioeconomic status (~7%). There was no evidence of an interaction between EduYears GPS and family socioeconomic status on educational achievement or on general cognitive ability. These results are a harbinger of future widespread use of GPS to predict genetic risk and resilience in the social and behavioral sciences.
Link
I wonder when the public and policymakers will get wind of the fact that educational achievement is highly heritable and can even be somewhat predicted with existing DNA technology.
Genetic egalitarianism is an edifice on which too much has been invested and I doubt that it will go down without a fight. It's of course a great idea to optimize learning for the students you've got. But, at the end of the day there's only so much you can do to foster achievement in a trait that is mostly genetically determined.
Molecular Psychiatry advance online publication 19 July 2016; doi: 10.1038/mp.2016.107
Predicting educational achievement from DNA
S Selzam et al.
A genome-wide polygenic score (GPS), derived from a 2013 genome-wide association study (N=127,000), explained 2% of the variance in total years of education (EduYears). In a follow-up study (N=329,000), a new EduYears GPS explains up to 4%. Here, we tested the association between this latest EduYears GPS and educational achievement scores at ages 7, 12 and 16 in an independent sample of 5825 UK individuals. We found that EduYears GPS explained greater amounts of variance in educational achievement over time, up to 9% at age 16, accounting for 15% of the heritable variance. This is the strongest GPS prediction to date for quantitative behavioral traits. Individuals in the highest and lowest GPS septiles differed by a whole school grade at age 16. Furthermore, EduYears GPS was associated with general cognitive ability (~3.5%) and family socioeconomic status (~7%). There was no evidence of an interaction between EduYears GPS and family socioeconomic status on educational achievement or on general cognitive ability. These results are a harbinger of future widespread use of GPS to predict genetic risk and resilience in the social and behavioral sciences.
Link
July 11, 2016
Y-chromosome haplogroup N phylogeny resolved
AJHG Volume 99, Issue 1, p163–173, 7 July 2016
Human Y Chromosome Haplogroup N: A Non-trivial Time-Resolved Phylogeography that Cuts across Language Families
Anne-Mai Ilumäe et al.
The paternal haplogroup (hg) N is distributed from southeast Asia to eastern Europe. The demographic processes that have shaped the vast extent of this major Y chromosome lineage across numerous linguistically and autosomally divergent populations have previously been unresolved. On the basis of 94 high-coverage re-sequenced Y chromosomes, we establish and date a detailed hg N phylogeny. We evaluate geographic structure by using 16 distinguishing binary markers in 1,631 hg N Y chromosomes from a collection of 6,521 samples from 56 populations. The more southerly distributed sub-clade N4 emerged before N2a1 and N3, found mostly in the north, but the latter two display more elaborate branching patterns, indicative of regional contrasts in recent expansions. In particular, a number of prominent and well-defined clades with common N3a3’6 ancestry occur in regionally dissimilar northern Eurasian populations, indicating almost simultaneous regional diversification and expansion within the last 5,000 years. This patrilineal genetic affinity is decoupled from the associated higher degree of language diversity.
Link
Human Y Chromosome Haplogroup N: A Non-trivial Time-Resolved Phylogeography that Cuts across Language Families
Anne-Mai Ilumäe et al.
The paternal haplogroup (hg) N is distributed from southeast Asia to eastern Europe. The demographic processes that have shaped the vast extent of this major Y chromosome lineage across numerous linguistically and autosomally divergent populations have previously been unresolved. On the basis of 94 high-coverage re-sequenced Y chromosomes, we establish and date a detailed hg N phylogeny. We evaluate geographic structure by using 16 distinguishing binary markers in 1,631 hg N Y chromosomes from a collection of 6,521 samples from 56 populations. The more southerly distributed sub-clade N4 emerged before N2a1 and N3, found mostly in the north, but the latter two display more elaborate branching patterns, indicative of regional contrasts in recent expansions. In particular, a number of prominent and well-defined clades with common N3a3’6 ancestry occur in regionally dissimilar northern Eurasian populations, indicating almost simultaneous regional diversification and expansion within the last 5,000 years. This patrilineal genetic affinity is decoupled from the associated higher degree of language diversity.
Link
June 27, 2016
37,000 year old skull from Malaysia related to indigenous people of Borneo
Front. Ecol. Evol., 27 June 2016 | http://dx.doi.org/10.3389/fevo.2016.00075
Deep Skull from Niah Cave and the Pleistocene Peopling of Southeast Asia
Darren Curnoe et al.
The Deep Skull from Niah Cave in Sarawak (Malaysia) is the oldest anatomically modern human recovered from island Southeast Asia. For more than 50 years its relevance to tracing the prehistory of the region has been controversial. The most widely held view, originating with Brothwell's 1960 description and analysis, is that the Niah individual is related to Indigenous Australians. Here we undertake a new assessment of the Deep Skull and consider its bearing on this question. In doing so, we provide a new and comprehensive description of the cranium including a reassessment of its ontogenetic age, sex, morphology, and affinities. We conclude that this individual was most likely to have been of advanced age and female, rather than an adolescent male as originally proposed. The morphological evidence strongly suggests that the Deep Skull samples the earliest modern humans to have settled Borneo, most likely originating on mainland East Asia. We also show that the affinities of the specimen are most likely to be with the contemporary indigenous people of Borneo, although, similarities to the population sometimes referred to as Philippine Negritos cannot be excluded. Finally, our research suggests that the widely supported “two-layer” hypothesis for the Pleistocene peopling of East/Southeast Asia is unlikely to apply to the earliest inhabitants of Borneo, in-line with the picture emerging from genetic studies of the contemporary people from the region.
Link
Deep Skull from Niah Cave and the Pleistocene Peopling of Southeast Asia
Darren Curnoe et al.
The Deep Skull from Niah Cave in Sarawak (Malaysia) is the oldest anatomically modern human recovered from island Southeast Asia. For more than 50 years its relevance to tracing the prehistory of the region has been controversial. The most widely held view, originating with Brothwell's 1960 description and analysis, is that the Niah individual is related to Indigenous Australians. Here we undertake a new assessment of the Deep Skull and consider its bearing on this question. In doing so, we provide a new and comprehensive description of the cranium including a reassessment of its ontogenetic age, sex, morphology, and affinities. We conclude that this individual was most likely to have been of advanced age and female, rather than an adolescent male as originally proposed. The morphological evidence strongly suggests that the Deep Skull samples the earliest modern humans to have settled Borneo, most likely originating on mainland East Asia. We also show that the affinities of the specimen are most likely to be with the contemporary indigenous people of Borneo, although, similarities to the population sometimes referred to as Philippine Negritos cannot be excluded. Finally, our research suggests that the widely supported “two-layer” hypothesis for the Pleistocene peopling of East/Southeast Asia is unlikely to apply to the earliest inhabitants of Borneo, in-line with the picture emerging from genetic studies of the contemporary people from the region.
Link
June 24, 2016
Population history with physically phased genomes
bioRxiv doi: http://dx.doi.org/10.1101/008367
Modeling human population separation history using physically phased genomes
Shiya Song, Elzbieta Sliwerska, Sarah Emery, Jeffrey M Kidd
Phased haplotype sequences are a key component in many population genetic analyses since variation in haplotypes reflects the action of recombination, selection, and changes in population size. In humans, haplotypes are typically estimated from unphased sequence or genotyping data using statistical models applied to large reference panels. To assess the importance of correct haplotype phase on population history inference, we performed fosmid pool sequencing and resolved phased haplotypes of five individuals from diverse African populations (including Yoruba, Esan, Gambia, Massai and Mende). We physically phased 98% of heterozygous SNPs into haplotype-resolved blocks, obtaining a block N50 of 1 Mbp. We combined these data with additional phased genomes from San, Mbuti, Gujarati and CEPH European populations and analyzed population size and separation history using the Pairwise Sequentially Markovian Coalescent (PSMC) and Multiple Sequentially Markovian Coalescent (MSMC) models. We find that statistically phased haplotypes yield an earlier split-time estimation compared with experimentally phased haplotypes. To better interpret patterns of cross-population coalescence, we implemented an approximate Bayesian computation (ABC) approach to estimate population split times and migration rates by fitting the distribution of coalescent times inferred between two haplotypes, one from each population, to a standard Isolation-with-Migration model. We inferred that the separation between hunter-gather populations and other populations happened around 120,000 to 140,000 years ago with gene flow continuing until 30,000 to 40,000 years ago; separation between west African and out of African populations happened around 70,000 to 80,000 years ago, while the separation between Massai and out of African populations happened around 50,000 years ago.
Link
Modeling human population separation history using physically phased genomes
Shiya Song, Elzbieta Sliwerska, Sarah Emery, Jeffrey M Kidd
Phased haplotype sequences are a key component in many population genetic analyses since variation in haplotypes reflects the action of recombination, selection, and changes in population size. In humans, haplotypes are typically estimated from unphased sequence or genotyping data using statistical models applied to large reference panels. To assess the importance of correct haplotype phase on population history inference, we performed fosmid pool sequencing and resolved phased haplotypes of five individuals from diverse African populations (including Yoruba, Esan, Gambia, Massai and Mende). We physically phased 98% of heterozygous SNPs into haplotype-resolved blocks, obtaining a block N50 of 1 Mbp. We combined these data with additional phased genomes from San, Mbuti, Gujarati and CEPH European populations and analyzed population size and separation history using the Pairwise Sequentially Markovian Coalescent (PSMC) and Multiple Sequentially Markovian Coalescent (MSMC) models. We find that statistically phased haplotypes yield an earlier split-time estimation compared with experimentally phased haplotypes. To better interpret patterns of cross-population coalescence, we implemented an approximate Bayesian computation (ABC) approach to estimate population split times and migration rates by fitting the distribution of coalescent times inferred between two haplotypes, one from each population, to a standard Isolation-with-Migration model. We inferred that the separation between hunter-gather populations and other populations happened around 120,000 to 140,000 years ago with gene flow continuing until 30,000 to 40,000 years ago; separation between west African and out of African populations happened around 70,000 to 80,000 years ago, while the separation between Massai and out of African populations happened around 50,000 years ago.
Link
June 21, 2016
Panorama of African admixture

Well, I won't dismiss the role of climate altogether, but it's hard to argue for it much anymore now that we know that the two big fish in the African ocean of human diversity were the spread of Niger-Congo languages (from the west), and of Caucasoids (from the east) over the last few thousands of years, with a healthy seasoning of minor admixtures before and after. Once again it seems that old-style anthropology was right and the more fashionable and trendy attempts to dismiss it as "typology", "imposition of European colonialism through science" and the like were wrong.
eLife 2016;5:e15266
Admixture into and within sub-Saharan Africa
George BJ Busby et al.
Similarity between two individuals in the combination of genetic markers along their chromosomes indicates shared ancestry and can be used to identify historical connections between different population groups due to admixture. We use a genome-wide, haplotype-based, analysis to characterise the structure of genetic diversity and gene-flow in a collection of 48 sub-Saharan African groups. We show that coastal populations experienced an influx of Eurasian haplotypes over the last 7000 years, and that Eastern and Southern Niger-Congo speaking groups share ancestry with Central West Africans as a result of recent population expansions. In fact, most sub-Saharan populations share ancestry with groups from outside of their current geographic region as a result of gene-flow within the last 4000 years. Our in-depth analysis provides insight into haplotype sharing across different ethno-linguistic groups and the recent movement of alleles into new environments, both of which are relevant to studies of genetic epidemiology.
Link
June 08, 2016
700 thousand year old ancestors of H. floresiensis
Nature 534, 245–248 (09 June 2016) doi:10.1038/nature17999
Homo floresiensis-like fossils from the early Middle Pleistocene of Flores
Gerrit D. van den Bergh, Yousuke Kaifu, Iwan Kurniawan, Reiko T. Kono, Adam Brumm, Erick Setiyabudi, Fachroel Aziz & Michael J. Morwood
The evolutionary origin of Homo floresiensis, a diminutive hominin species previously known only by skeletal remains from Liang Bua in western Flores, Indonesia, has been intensively debated. It is a matter of controversy whether this primitive form, dated to the Late Pleistocene, evolved from early Asian Homo erectus and represents a unique and striking case of evolutionary reversal in hominin body and brain size within an insular environment1, 2, 3, 4. The alternative hypothesis is that H. floresiensis derived from an older, smaller-brained member of our genus, such as Homo habilis, or perhaps even late Australopithecus, signalling a hitherto undocumented dispersal of hominins from Africa into eastern Asia by two million years ago (2 Ma)5, 6. Here we describe hominin fossils excavated in 2014 from an early Middle Pleistocene site (Mata Menge) in the So’a Basin of central Flores. These specimens comprise a mandible fragment and six isolated teeth belonging to at least three small-jawed and small-toothed individuals. Dating to ~0.7 Ma, these fossils now constitute the oldest hominin remains from Flores7. The Mata Menge mandible and teeth are similar in dimensions and morphological characteristics to those of H. floresiensis from Liang Bua. The exception is the mandibular first molar, which retains a more primitive condition. Notably, the Mata Menge mandible and molar are even smaller in size than those of the two existing H. floresiensis individuals from Liang Bua. The Mata Menge fossils are derived compared with Australopithecus and H. habilis, and so tend to support the view that H. floresiensis is a dwarfed descendent of early Asian H. erectus. Our findings suggest that hominins on Flores had acquired extremely small body size and other morphological traits specific to H. floresiensis at an unexpectedly early time.
Link
Nature 534, 249–253 (09 June 2016) doi:10.1038/nature17663
Age and context of the oldest known hominin fossils from Flores
Adam Brumm, Gerrit D. van den Bergh, Michael Storey, Iwan Kurniawan, Brent V. Alloway, Ruly Setiawan, Erick Setiyabudi, Rainer Grün, Mark W. Moore, Dida Yurnaldi, Mika R. Puspaningrum, Unggul P. Wibowo, Halmi Insani, Indra Sutisna, John A. Westgate, Nick J. G. Pearce, Mathieu Duval, Hanneke J. M. Meijer, Fachroel Aziz, Thomas Sutikna, Sander van der Kaars, Stephanie Flude & Michael J. Morwood
Recent excavations at the early Middle Pleistocene site of Mata Menge in the So’a Basin of central Flores, Indonesia, have yielded hominin fossils1 attributed to a population ancestral to Late Pleistocene Homo floresiensis2. Here we describe the age and context of the Mata Menge hominin specimens and associated archaeological findings. The fluvial sandstone layer from which the in situ fossils were excavated in 2014 was deposited in a small valley stream around 700 thousand years ago, as indicated by 40Ar/39Ar and fission track dates on stratigraphically bracketing volcanic ash and pyroclastic density current deposits, in combination with coupled uranium-series and electron spin resonance dating of fossil teeth. Palaeoenvironmental data indicate a relatively dry climate in the So’a Basin during the early Middle Pleistocene, while various lines of evidence suggest the hominins inhabited a savannah-like open grassland habitat with a wetland component. The hominin fossils occur alongside the remains of an insular fauna and a simple stone technology that is markedly similar to that associated with Late Pleistocene H. floresiensis.
Link
Homo floresiensis-like fossils from the early Middle Pleistocene of Flores
Gerrit D. van den Bergh, Yousuke Kaifu, Iwan Kurniawan, Reiko T. Kono, Adam Brumm, Erick Setiyabudi, Fachroel Aziz & Michael J. Morwood
The evolutionary origin of Homo floresiensis, a diminutive hominin species previously known only by skeletal remains from Liang Bua in western Flores, Indonesia, has been intensively debated. It is a matter of controversy whether this primitive form, dated to the Late Pleistocene, evolved from early Asian Homo erectus and represents a unique and striking case of evolutionary reversal in hominin body and brain size within an insular environment1, 2, 3, 4. The alternative hypothesis is that H. floresiensis derived from an older, smaller-brained member of our genus, such as Homo habilis, or perhaps even late Australopithecus, signalling a hitherto undocumented dispersal of hominins from Africa into eastern Asia by two million years ago (2 Ma)5, 6. Here we describe hominin fossils excavated in 2014 from an early Middle Pleistocene site (Mata Menge) in the So’a Basin of central Flores. These specimens comprise a mandible fragment and six isolated teeth belonging to at least three small-jawed and small-toothed individuals. Dating to ~0.7 Ma, these fossils now constitute the oldest hominin remains from Flores7. The Mata Menge mandible and teeth are similar in dimensions and morphological characteristics to those of H. floresiensis from Liang Bua. The exception is the mandibular first molar, which retains a more primitive condition. Notably, the Mata Menge mandible and molar are even smaller in size than those of the two existing H. floresiensis individuals from Liang Bua. The Mata Menge fossils are derived compared with Australopithecus and H. habilis, and so tend to support the view that H. floresiensis is a dwarfed descendent of early Asian H. erectus. Our findings suggest that hominins on Flores had acquired extremely small body size and other morphological traits specific to H. floresiensis at an unexpectedly early time.
Link
Nature 534, 249–253 (09 June 2016) doi:10.1038/nature17663
Age and context of the oldest known hominin fossils from Flores
Adam Brumm, Gerrit D. van den Bergh, Michael Storey, Iwan Kurniawan, Brent V. Alloway, Ruly Setiawan, Erick Setiyabudi, Rainer Grün, Mark W. Moore, Dida Yurnaldi, Mika R. Puspaningrum, Unggul P. Wibowo, Halmi Insani, Indra Sutisna, John A. Westgate, Nick J. G. Pearce, Mathieu Duval, Hanneke J. M. Meijer, Fachroel Aziz, Thomas Sutikna, Sander van der Kaars, Stephanie Flude & Michael J. Morwood
Recent excavations at the early Middle Pleistocene site of Mata Menge in the So’a Basin of central Flores, Indonesia, have yielded hominin fossils1 attributed to a population ancestral to Late Pleistocene Homo floresiensis2. Here we describe the age and context of the Mata Menge hominin specimens and associated archaeological findings. The fluvial sandstone layer from which the in situ fossils were excavated in 2014 was deposited in a small valley stream around 700 thousand years ago, as indicated by 40Ar/39Ar and fission track dates on stratigraphically bracketing volcanic ash and pyroclastic density current deposits, in combination with coupled uranium-series and electron spin resonance dating of fossil teeth. Palaeoenvironmental data indicate a relatively dry climate in the So’a Basin during the early Middle Pleistocene, while various lines of evidence suggest the hominins inhabited a savannah-like open grassland habitat with a wetland component. The hominin fossils occur alongside the remains of an insular fauna and a simple stone technology that is markedly similar to that associated with Late Pleistocene H. floresiensis.
Link
June 07, 2016
Neolithic Aegean genomes


PNAS doi: 10.1073/pnas.1523951113
Early farmers from across Europe directly descended from Neolithic Aegeans
Zuzana Hofmanová, Susanne Kreutzer et al.
Farming and sedentism first appeared in southwestern Asia during the early Holocene and later spread to neighboring regions, including Europe, along multiple dispersal routes. Conspicuous uncertainties remain about the relative roles of migration, cultural diffusion, and admixture with local foragers in the early Neolithization of Europe. Here we present paleogenomic data for five Neolithic individuals from northern Greece and northwestern Turkey spanning the time and region of the earliest spread of farming into Europe. We use a novel approach to recalibrate raw reads and call genotypes from ancient DNA and observe striking genetic similarity both among Aegean early farmers and with those from across Europe. Our study demonstrates a direct genetic link between Mediterranean and Central European early farmers and those of Greece and Anatolia, extending the European Neolithic migratory chain all the way back to southwestern Asia.
Link
Ancient DNA and human history

The lack of East Asian DNA validates my New Year's wish for some. Hopefully my wish will be granted in the second half of 2016.
PNAS doi: 10.1073/pnas.1524306113
Ancient DNA and human history
Montgomery Slatkin, and Fernando Racimo
We review studies of genomic data obtained by sequencing hominin fossils with particular emphasis on the unique information that ancient DNA (aDNA) can provide about the demographic history of humans and our closest relatives. We concentrate on nuclear genomic sequences that have been published in the past few years. In many cases, particularly in the Arctic, the Americas, and Europe, aDNA has revealed historical demographic patterns in a way that could not be resolved by analyzing present-day genomes alone. Ancient DNA from archaic hominins has revealed a rich history of admixture between early modern humans, Neanderthals, and Denisovans, and has allowed us to disentangle complex selective processes. Information from aDNA studies is nowhere near saturation, and we believe that future aDNA sequences will continue to change our understanding of hominin history.
Link
Mungo Man DNA revisited + first ancient mtDNA from Australia
The authors find that previously published mtDNA from earliest Australians was contamination, and one S2 mtDNA haplogroup in an undated sample of likely Holocene origin.
PNAS doi: 10.1073/pnas.1521066113
Ancient mtDNA sequences from the First Australians revisited
Tim H. Heupink et al.
The publication in 2001 by Adcock et al. [Adcock GJ, et al. (2001) Proc Natl Acad Sci USA 98(2):537–542] in PNAS reported the recovery of short mtDNA sequences from ancient Australians, including the 42,000-y-old Mungo Man [Willandra Lakes Hominid (WLH3)]. This landmark study in human ancient DNA suggested that an early modern human mitochondrial lineage emerged in Asia and that the theory of modern human origins could no longer be considered solely through the lens of the “Out of Africa” model. To evaluate these claims, we used second generation DNA sequencing and capture methods as well as PCR-based and single-primer extension (SPEX) approaches to reexamine the same four Willandra Lakes and Kow Swamp 8 (KS8) remains studied in the work by Adcock et al. Two of the remains sampled contained no identifiable human DNA (WLH15 and WLH55), whereas the Mungo Man (WLH3) sample contained no Aboriginal Australian DNA. KS8 reveals human mitochondrial sequences that differ from the previously inferred sequence. Instead, we recover a total of five modern European contaminants from Mungo Man (WLH3). We show that the remaining sample (WLH4) contains ∼1.4% human DNA, from which we assembled two complete mitochondrial genomes. One of these was a previously unidentified Aboriginal Australian haplotype belonging to haplogroup S2 that we sequenced to a high coverage. The other was a contaminating modern European mitochondrial haplotype. Although none of the sequences that we recovered matched those reported by Adcock et al., except a contaminant, these findings show the feasibility of obtaining important information from ancient Aboriginal Australian remains.
Link
PNAS doi: 10.1073/pnas.1521066113
Ancient mtDNA sequences from the First Australians revisited
Tim H. Heupink et al.
The publication in 2001 by Adcock et al. [Adcock GJ, et al. (2001) Proc Natl Acad Sci USA 98(2):537–542] in PNAS reported the recovery of short mtDNA sequences from ancient Australians, including the 42,000-y-old Mungo Man [Willandra Lakes Hominid (WLH3)]. This landmark study in human ancient DNA suggested that an early modern human mitochondrial lineage emerged in Asia and that the theory of modern human origins could no longer be considered solely through the lens of the “Out of Africa” model. To evaluate these claims, we used second generation DNA sequencing and capture methods as well as PCR-based and single-primer extension (SPEX) approaches to reexamine the same four Willandra Lakes and Kow Swamp 8 (KS8) remains studied in the work by Adcock et al. Two of the remains sampled contained no identifiable human DNA (WLH15 and WLH55), whereas the Mungo Man (WLH3) sample contained no Aboriginal Australian DNA. KS8 reveals human mitochondrial sequences that differ from the previously inferred sequence. Instead, we recover a total of five modern European contaminants from Mungo Man (WLH3). We show that the remaining sample (WLH4) contains ∼1.4% human DNA, from which we assembled two complete mitochondrial genomes. One of these was a previously unidentified Aboriginal Australian haplotype belonging to haplogroup S2 that we sequenced to a high coverage. The other was a contaminating modern European mitochondrial haplotype. Although none of the sequences that we recovered matched those reported by Adcock et al., except a contaminant, these findings show the feasibility of obtaining important information from ancient Aboriginal Australian remains.
Link
May 28, 2016
British Celts have more steppe ancestry than British English
An interesting tidbit in a preprint about blood pressure genes:
But, it seems that the English have less steppe ancestry than both modern Celts and ancient Saxons, so they're not really intermediate. My guess is that the English have Norman ancestry that the Celts don't. While the original Normans were Scandinavians with presumably lots of steppe ancestry, I'd be surprised if the post-1066 Normans that settled England were not already heavily admixed with the "French" and so had less steppe ancestry than the modern British Celts from Wales and Scotland.
bioRxiv http://dx.doi.org/10.1101/055855
Population structure of UK Biobank and ancient Eurasians reveals adaptation at genes influencing blood pressure
Kevin Galinsky et al.
Analyzing genetic differences between closely related populations can be a powerful way to detect recent adaptation. The very large sample size of the UK Biobank is ideal for detecting selection using population differentiation, and enables an analysis of UK population structure at fine resolution. In analyses of 113,851 UK Biobank samples, population structure in the UK is dominated by 5 principal components (PCs) spanning 6 clusters: Northern Ireland, Scotland, northern England, southern England, and two Welsh clusters. Analyses with ancient Eurasians show that populations in the northern UK have higher levels of Steppe ancestry, and that UK population structure cannot be explained as a simple mixture of Celts and Saxons. A scan for unusual population differentiation along top PCs identified a genome-wide significant signal of selection at the coding variant rs601338 in FUT2 (p=9.16×10-9). In addition, by combining evidence of unusual differentiation within the UK with evidence from ancient Eurasians, we identified new genome-wide significant (p less than 5×10-8) signals of recent selection at two additional loci: CYP1A2/CSK and F12. We detected strong associations to diastolic blood pressure in the UK Biobank for the variants with new selection signals at CYP1A2/CSK (p=1.10×10-19)) and for variants with ancient Eurasian selection signals in the ATXN2/SH2B3 locus (p=8.00×10-33), implicating recent adaptation related to blood pressure.
Link
We consistently obtained significantly positive f4 statistics, implying that both the modern Celtic samples and the ancient Saxon samples have more Steppe ancestry than the modern Anglo-Saxon samples from southern and eastern England. This indicates that southern and eastern England is not exclusively a genetic mix of Celts and Saxons.Southeastern England is genetically very homogeneous. If the people there were a mix of ancient Celts and Saxons you'd expect them to be intermediate between modern Celts (who should have more Celtic ancestry than the modern English) and ancient Saxons (who should have more Saxon ancestry than the modern English).
But, it seems that the English have less steppe ancestry than both modern Celts and ancient Saxons, so they're not really intermediate. My guess is that the English have Norman ancestry that the Celts don't. While the original Normans were Scandinavians with presumably lots of steppe ancestry, I'd be surprised if the post-1066 Normans that settled England were not already heavily admixed with the "French" and so had less steppe ancestry than the modern British Celts from Wales and Scotland.
bioRxiv http://dx.doi.org/10.1101/055855
Population structure of UK Biobank and ancient Eurasians reveals adaptation at genes influencing blood pressure
Kevin Galinsky et al.
Analyzing genetic differences between closely related populations can be a powerful way to detect recent adaptation. The very large sample size of the UK Biobank is ideal for detecting selection using population differentiation, and enables an analysis of UK population structure at fine resolution. In analyses of 113,851 UK Biobank samples, population structure in the UK is dominated by 5 principal components (PCs) spanning 6 clusters: Northern Ireland, Scotland, northern England, southern England, and two Welsh clusters. Analyses with ancient Eurasians show that populations in the northern UK have higher levels of Steppe ancestry, and that UK population structure cannot be explained as a simple mixture of Celts and Saxons. A scan for unusual population differentiation along top PCs identified a genome-wide significant signal of selection at the coding variant rs601338 in FUT2 (p=9.16×10-9). In addition, by combining evidence of unusual differentiation within the UK with evidence from ancient Eurasians, we identified new genome-wide significant (p less than 5×10-8) signals of recent selection at two additional loci: CYP1A2/CSK and F12. We detected strong associations to diastolic blood pressure in the UK Biobank for the variants with new selection signals at CYP1A2/CSK (p=1.10×10-19)) and for variants with ancient Eurasian selection signals in the ATXN2/SH2B3 locus (p=8.00×10-33), implicating recent adaptation related to blood pressure.
Link
May 27, 2016
The great migration of African Americans

The Great Migration and African-American Genomic Diversity
Soheil Baharian et al.
We present a comprehensive assessment of genomic diversity in the African-American population by studying three genotyped cohorts comprising 3,726 African-Americans from across the United States that provide a representative description of the population across all US states and socioeconomic status. An estimated 82.1% of ancestors to African-Americans lived in Africa prior to the advent of transatlantic travel, 16.7% in Europe, and 1.2% in the Americas, with increased African ancestry in the southern United States compared to the North and West. Combining demographic models of ancestry and those of relatedness suggests that admixture occurred predominantly in the South prior to the Civil War and that ancestry-biased migration is responsible for regional differences in ancestry. We find that recent migrations also caused a strong increase in genetic relatedness among geographically distant African-Americans. Long-range relatedness among African-Americans and between African-Americans and European-Americans thus track north- and west-bound migration routes followed during the Great Migration of the twentieth century. By contrast, short-range relatedness patterns suggest comparable mobility of ∼15–16km per generation for African-Americans and European-Americans, as estimated using a novel analytical model of isolation-by-distance.
Link
May 19, 2016
35,000 year old mtDNA haplogroup U6 from Romania
I wouldn't be very surprised if many of the markers supposedly signifying recent gene flow Africa and Eurasia were actually quite old in Eurasia. The trouble is that reports of such gene flow were often based on simply observing that marker "X" occurs at a higher frequency in Africa than in Eurasia, so a common sense explanation is that it reflects limited recent gene flow between the continents. But, it is now known that common sense is not always the best guide, as e.g., ancient Europeans had mtDNA haplogroup M (in the past considered evidence of Asian admixture), Y-chromosome haplogroup C (ditto), and now U6.
The same should also apply to the Middle East where there has been admixture with Africans since the Islamic period at least. The existence of such admixture does not mean that every single lineage that occurs at low frequency in the Middle East and high frequency in Africa is diagnostic of this later period of admixture. Some of them could well be relics of old Middle Eastern populations. Who knows what people inhabited the presently inhospitable landscape of the Saharan-Arabian desert zone? The living populations can certainly make no claim to being the first ones there, but the genetic heritage of those earlier occupants may still persist in them in traces.
Similarly for the New World; in that case, there is a better case that European-looking lineages are indeed due to the colonization of the Americas over the last five centuries. However, that does not mean that all of them are, and we should be mindful of the possibility of pre-Columbian contact between the Old and New worlds.
Scientific Reports 6, Article number: 25501 (2016)
The mitogenome of a 35,000-year-old Homo sapiens from Europe supports a Palaeolithic back-migration to Africa
M. Hervella et al.
After the dispersal of modern humans (Homo sapiens) Out of Africa, hominins with a similar morphology to that of present-day humans initiated the gradual demographic expansion into Eurasia. The mitogenome (33-fold coverage) of the Peştera Muierii 1 individual (PM1) from Romania (35 ky cal BP) we present in this article corresponds fully to Homo sapiens, whilst exhibiting a mosaic of morphological features related to both modern humans and Neandertals. We have identified the PM1 mitogenome as a basal haplogroup U6*, not previously found in any ancient or present-day humans. The derived U6 haplotypes are predominantly found in present-day North-Western African populations. Concomitantly, those found in Europe have been attributed to recent gene-flow from North Africa. The presence of the basal haplogroup U6* in South East Europe (Romania) at 35 ky BP confirms a Eurasian origin of the U6 mitochondrial lineage. Consequently, we propose that the PM1 lineage is an offshoot to South East Europe that can be traced to the Early Upper Paleolithic back migration from Western Asia to North Africa, during which the U6 lineage diversified, until the emergence of the present-day U6 African lineages.
Link
The same should also apply to the Middle East where there has been admixture with Africans since the Islamic period at least. The existence of such admixture does not mean that every single lineage that occurs at low frequency in the Middle East and high frequency in Africa is diagnostic of this later period of admixture. Some of them could well be relics of old Middle Eastern populations. Who knows what people inhabited the presently inhospitable landscape of the Saharan-Arabian desert zone? The living populations can certainly make no claim to being the first ones there, but the genetic heritage of those earlier occupants may still persist in them in traces.
Similarly for the New World; in that case, there is a better case that European-looking lineages are indeed due to the colonization of the Americas over the last five centuries. However, that does not mean that all of them are, and we should be mindful of the possibility of pre-Columbian contact between the Old and New worlds.
Scientific Reports 6, Article number: 25501 (2016)
The mitogenome of a 35,000-year-old Homo sapiens from Europe supports a Palaeolithic back-migration to Africa
M. Hervella et al.
After the dispersal of modern humans (Homo sapiens) Out of Africa, hominins with a similar morphology to that of present-day humans initiated the gradual demographic expansion into Eurasia. The mitogenome (33-fold coverage) of the Peştera Muierii 1 individual (PM1) from Romania (35 ky cal BP) we present in this article corresponds fully to Homo sapiens, whilst exhibiting a mosaic of morphological features related to both modern humans and Neandertals. We have identified the PM1 mitogenome as a basal haplogroup U6*, not previously found in any ancient or present-day humans. The derived U6 haplotypes are predominantly found in present-day North-Western African populations. Concomitantly, those found in Europe have been attributed to recent gene-flow from North Africa. The presence of the basal haplogroup U6* in South East Europe (Romania) at 35 ky BP confirms a Eurasian origin of the U6 mitochondrial lineage. Consequently, we propose that the PM1 lineage is an offshoot to South East Europe that can be traced to the Early Upper Paleolithic back migration from Western Asia to North Africa, during which the U6 lineage diversified, until the emergence of the present-day U6 African lineages.
Link
May 12, 2016
Luwians vs. Hittites and Mycenaeans vs. Luwians

May 11, 2016
74 loci associated with educational attainment
Other than the claim in the abstract that educational attainment is "mostly environmentally determined" (*), this seems like a very useful study, as it identifies 74 loci associated with educational attainment and explores their interesting biology.
The utility of this type of study does not consist so much in the ability to predict one's educational potential by looking at one's genotype (we're a long way off from that, and a traditional pencil-and-paper test will probably beat genetics for a long time to come). Rather, it helps move the culture forward, away from the polite ultra-egalitarianism of today's dominant worldview and towards a more scientific attitude concerning the limits of education. Such an attitude will necessarily acknowledge -whether it seems fair to us or not- that genes sometimes dictate that the smart but slothful kid should outperform his diligent but dull-witted peer.
It will certainly be very interesting to see what better methods or even larger sample sizes will bring in years to come.
It will certainly be very interesting to see what better methods or even larger sample sizes will bring in years to come.
(*) The heritability of educational attainment has been estimated to be 67-74% of Norwegian males of the 1940-1961 period. There is actually no "universal heritability" of a trait. In a third world country it may very well be that one's educational attainment is determined mostly by environmental effects such as whether you have access to a school within reasonable distance or to just enough food during development. In a modern country (like post-war Norway or a technologically advanced future utopia), environmental effects are expected to be minimal (as everyone will get the best of everything), and variation in educational attainment will simply be due to genes (+noise).
Nature (2016) doi:10.1038/nature17671
Genome-wide association study identifies 74 loci associated with educational attainment
Aysu Okbay et al.
Educational attainment is strongly influenced by social and other environmental factors, but genetic factors are estimated to account for at least 20% of the variation across individuals1. Here we report the results of a genome-wide association study (GWAS) for educational attainment that extends our earlier discovery sample1, 2 of 101,069 individuals to 293,723 individuals, and a replication study in an independent sample of 111,349 individuals from the UK Biobank. We identify 74 genome-wide significant loci associated with the number of years of schooling completed. Single-nucleotide polymorphisms associated with educational attainment are disproportionately found in genomic regions regulating gene expression in the fetal brain. Candidate genes are preferentially expressed in neural tissue, especially during the prenatal period, and enriched for biological pathways involved in neural development. Our findings demonstrate that, even for a behavioural phenotype that is mostly environmentally determined, a well-powered GWAS identifies replicable associated genetic variants that suggest biologically relevant pathways. Because educational attainment is measured in large numbers of individuals, it will continue to be useful as a proxy phenotype in efforts to characterize the genetic influences of related phenotypes, including cognition and neuropsychiatric diseases.
Link
May 08, 2016
Natural selection in Britain during the last 2,000 years
The latest ancient DNA studies from the British Isles (Schiffels et al and Martiniano et al. and Cassidy et al.) support continuity over the last 2,000 years. Sure, there were continued migrations like the arrival of the Anglo-Saxons, but these were very similar groups in the grand scheme of things.
But, while ancestrally the modern Briton is probably a descendant of the Britons of 2,000 years ago with some admixture from similar continental European groups, he is not the same, as (apparently) substantial genetic adaptation has continued to operate in Britain over the same period. A new preprint by Field, Boyle, Telis et al. makes the case for adaptation in a variety of traits in the ancestors of Britons over this period. Mind you, the genetic underpinnings of many important human traits known to have high heritability are currently unknown, but there is little doubt that selection would have affected traits beyond those detected in this study. I am quite curious to see whether the striking efflorescence of cultural achievement in Britain over the last half millennium could have (at least in part) a genetic underpinning.
Depigmentation is a trait whose genetic architecture is as well as understood as any. The results of this study might surprise writers of decades and centuries past who supposed that the spectrum of pigmentation of modern Europeans was the result of admixture-in varying measure- between Xanthochrooi and Melanchrooi races of primordial antiquity. All indications seem to be that depigmentation of hair, skin, and eyes did not co-occur in such a hypothetical race, but rather in different parts of the Caucasoid range, only reaching a high combined frequency in northern Europe to form the distinctive physical type that is distinctive of the natives of that region. It would be quite interesting to see how these traits evolved in Fennoscandia and the Baltic, regions that sport an even higher depigmentation than the British Isles. Traditionally, these areas were viewed as refuges of the Xanthochrooi but it may very well turn out to be that for whatever reason selection has acted in that area as well, as it did in the Eastern European plain where rather dark Bronze Age steppe groups gave way to rather light pigmented living eastern Slavs.
bioRxiv doi: http://dx.doi.org/10.1101/052084
Detection of human adaptation during the past 2,000 years
Yair Field, Evan A Boyle, Natalie Telis, Ziyue Gao, Kyle J Gaulton, David Golan, Loic Yengo, Ghislain Rocheleau, Philippe Froguel, Mark I McCarthy, Jonathan K Pritchard
Detection of recent natural selection is a challenging problem in population genetics, as standard methods generally integrate over long timescales. Here we introduce the Singleton Density Score (SDS), a powerful measure to infer very recent changes in allele frequencies from contemporary genome sequences. When applied to data from the UK10K Project, SDS reflects allele frequency changes in the ancestors of modern Britons during the past 2,000 years. We see strong signals of selection at lactase and HLA, and in favor of blond hair and blue eyes. Turning to signals of polygenic adaptation we find, remarkably, that recent selection for increased height has driven allele frequency shifts across most of the genome. Moreover, we report suggestive new evidence for polygenic shifts affecting many other complex traits. Our results suggest that polygenic adaptation has played a pervasive role in shaping genotypic and phenotypic variation in modern humans.
Link
But, while ancestrally the modern Briton is probably a descendant of the Britons of 2,000 years ago with some admixture from similar continental European groups, he is not the same, as (apparently) substantial genetic adaptation has continued to operate in Britain over the same period. A new preprint by Field, Boyle, Telis et al. makes the case for adaptation in a variety of traits in the ancestors of Britons over this period. Mind you, the genetic underpinnings of many important human traits known to have high heritability are currently unknown, but there is little doubt that selection would have affected traits beyond those detected in this study. I am quite curious to see whether the striking efflorescence of cultural achievement in Britain over the last half millennium could have (at least in part) a genetic underpinning.
Depigmentation is a trait whose genetic architecture is as well as understood as any. The results of this study might surprise writers of decades and centuries past who supposed that the spectrum of pigmentation of modern Europeans was the result of admixture-in varying measure- between Xanthochrooi and Melanchrooi races of primordial antiquity. All indications seem to be that depigmentation of hair, skin, and eyes did not co-occur in such a hypothetical race, but rather in different parts of the Caucasoid range, only reaching a high combined frequency in northern Europe to form the distinctive physical type that is distinctive of the natives of that region. It would be quite interesting to see how these traits evolved in Fennoscandia and the Baltic, regions that sport an even higher depigmentation than the British Isles. Traditionally, these areas were viewed as refuges of the Xanthochrooi but it may very well turn out to be that for whatever reason selection has acted in that area as well, as it did in the Eastern European plain where rather dark Bronze Age steppe groups gave way to rather light pigmented living eastern Slavs.
bioRxiv doi: http://dx.doi.org/10.1101/052084
Detection of human adaptation during the past 2,000 years
Yair Field, Evan A Boyle, Natalie Telis, Ziyue Gao, Kyle J Gaulton, David Golan, Loic Yengo, Ghislain Rocheleau, Philippe Froguel, Mark I McCarthy, Jonathan K Pritchard
Detection of recent natural selection is a challenging problem in population genetics, as standard methods generally integrate over long timescales. Here we introduce the Singleton Density Score (SDS), a powerful measure to infer very recent changes in allele frequencies from contemporary genome sequences. When applied to data from the UK10K Project, SDS reflects allele frequency changes in the ancestors of modern Britons during the past 2,000 years. We see strong signals of selection at lactase and HLA, and in favor of blond hair and blue eyes. Turning to signals of polygenic adaptation we find, remarkably, that recent selection for increased height has driven allele frequency shifts across most of the genome. Moreover, we report suggestive new evidence for polygenic shifts affecting many other complex traits. Our results suggest that polygenic adaptation has played a pervasive role in shaping genotypic and phenotypic variation in modern humans.
Link
May 02, 2016
Neandertal ancestry, going, going, ..., gone (?)

The authors write:
Using one statistic, we estimate a decline from 4.3–5.7% from a time shortly after introgression to 1.1–2.2% in Eurasians today (Fig. 2).This is remarkable because it shows that most of the Neandertal ancestry of the earliest AMH in Europe was gone by the Mesolithic. It really seems that Neandertal genes were bred out of the gene pool over time. Will this trend continue into the future? Perhaps only minute traces of Neandertal DNA will remain in humans in 10,000 more years. Some of Neandertal DNA may yet prove to be neutral or beneficial, so at the limit the percentage may be more than zero. Nonetheless, the historical trend does suggest that modern humans inherited mostly genetic garbage from Neandertals and evolution is more than halfway through the process of getting rid of it.
As a corollary, there may have been other episodes of archaic admixture that are no longer detectable. Perhaps our modern human lineage has repeatedly admixed with other species, but traces of those admixtures are long gone by the action of natural selection. The reason for our relative homogeneity as a species may not be that we avoided intermixing with others, but that, sadly, most others had not much that was beneficial to offer to our ancestors.
Nature (2016) doi:10.1038/nature17993
The genetic history of Ice Age Europe
Qiaomei Fu et al.
Modern humans arrived in Europe ~45,000 years ago, but little is known about their genetic composition before the start of farming ~8,500 years ago. Here we analyse genome-wide data from 51 Eurasians from ~45,000–7,000 years ago. Over this time, the proportion of Neanderthal DNA decreased from 3–6% to around 2%, consistent with natural selection against Neanderthal variants in modern humans. Whereas there is no evidence of the earliest modern humans in Europe contributing to the genetic composition of present-day Europeans, all individuals between ~37,000 and ~14,000 years ago descended from a single founder population which forms part of the ancestry of present-day Europeans. An ~35,000-year-old individual from northwest Europe represents an early branch of this founder population which was then displaced across a broad region, before reappearing in southwest Europe at the height of the last Ice Age ~19,000 years ago. During the major warming period after ~14,000 years ago, a genetic component related to present-day Near Easterners became widespread in Europe. These results document how population turnover and migration have been recurring themes of European prehistory.
Link
May 01, 2016
April 30, 2016
More on Kennewick Man
A new technical report re-analyzes the data of Rasmussen et al. study on Kennewick man and confirms that he is related to Native Americans. From the report:

Technical Report: Assessment of the genetic analyses of Rasmussen et al. (2015)
John Novembre, PhD, David Witonsky, Anna Di Rienzo, PhD
The primary aim of the analysis undertaken here (U.S. Army Corps of Engineers, St Louis District Contract #W912P9-16-P-0010) is to provide an independent validation of the genetic evidence underlying a recent publication by Morten Rasmussen and colleagues on July 23rd, 2015, in Nature (Vol 523:455–58). Based on our analysis of the Kennewick Man’s sequence data and Colville tribe genotype data generated by Rasmussen et al., we concur with the findings of the original paper that the sample is genetically closer to modern Native Americans than to any other population worldwide. We carried out several analyses to support this conclusion, including (i) principal component analysis (PCA; Patterson et al. 2006), (ii) unsupervised genetic clustering using ADMIXTURE (Alexander, Novembre, and Lange 2009), (iii) estimation of genetic affinity to modern human populations using f3 and D statistics (Patterson et al. 2012), and (iv) a novel approach based on the geographic distribution of rare variants. Importantly, these distinct analyses, spanning three non-overlapping subsets of the data, are each consistent with Native American ancestry.
Link
We find the Kennewick sample has the highest shared similarity to Native American populations with the highest values observed being with populations from South America (Figure 7), in line with the observations from Rasmussen et al.Hopefully this will end the campaign to put him back to the ground. I have added a horizontal line to the new study's Figure 7 to mark the population claiming the skeleton among the huge number considered, showing that there's no particularly strong relationship to it (the strongest connection is at the bottom of the figure).
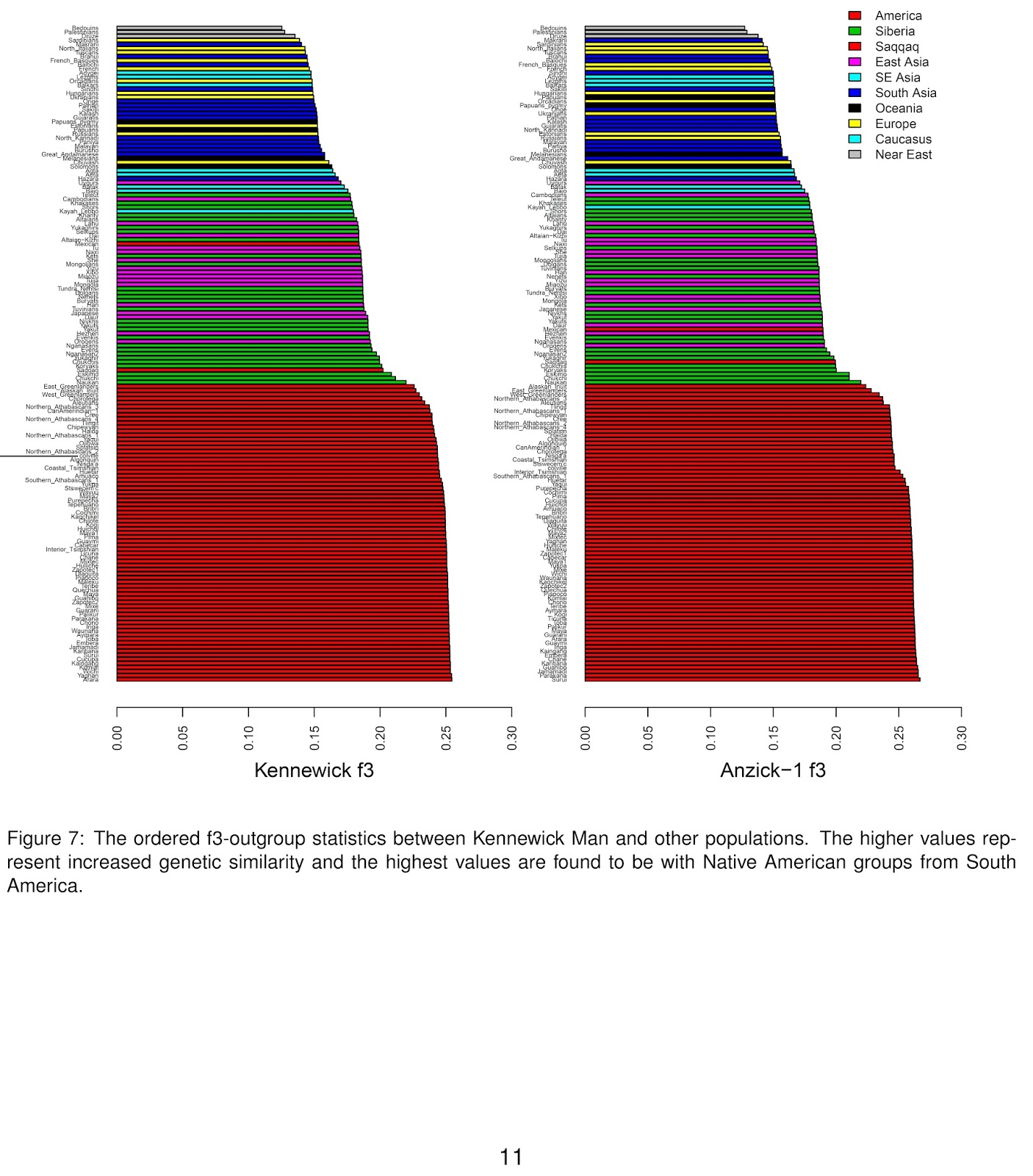
The Rasmussen et al. and Novembre et al. studies are really science working at its best: simultaneously falsifying claims that Kennewick was some sort of Australoid (or even more implausibly Caucasoid) based on its craniofacial morphology, but not overreaching to validate emotional appeals to make him into an ancestor he wasn't. Thankfully, the way forward is to keep studying Kennewick Man (and modern Native Americans) with ever-better data and techniques which may turn up (who knows?) a real (rather than imagined) ancestral link.
Technical Report: Assessment of the genetic analyses of Rasmussen et al. (2015)
John Novembre, PhD, David Witonsky, Anna Di Rienzo, PhD
The primary aim of the analysis undertaken here (U.S. Army Corps of Engineers, St Louis District Contract #W912P9-16-P-0010) is to provide an independent validation of the genetic evidence underlying a recent publication by Morten Rasmussen and colleagues on July 23rd, 2015, in Nature (Vol 523:455–58). Based on our analysis of the Kennewick Man’s sequence data and Colville tribe genotype data generated by Rasmussen et al., we concur with the findings of the original paper that the sample is genetically closer to modern Native Americans than to any other population worldwide. We carried out several analyses to support this conclusion, including (i) principal component analysis (PCA; Patterson et al. 2006), (ii) unsupervised genetic clustering using ADMIXTURE (Alexander, Novembre, and Lange 2009), (iii) estimation of genetic affinity to modern human populations using f3 and D statistics (Patterson et al. 2012), and (iv) a novel approach based on the geographic distribution of rare variants. Importantly, these distinct analyses, spanning three non-overlapping subsets of the data, are each consistent with Native American ancestry.
Link
April 25, 2016
Bursts in human male demography

When the tree is calibrated with a mutation rate estimate of 0.76 × 10-9 mutations per base pair per year9, the time to the most recent common ancestor (TMRCA) of the tree is ~190,000 years, but we consider the implications of alternative mutation rate estimates below. Of the clades resulting from the four deepest branching events, all but one are exclusive to Africa, and the TMRCA of all non-African lineages (that is, the TMRCA of haplogroups DE and CF) is ~76,000 years (Fig. 1, Supplementary Figs. 18 and 19, Supplementary Table 10, and Supplementary Note). We saw a notable increase in the number of lineages outside Africa ~50–55 kya, perhaps reflecting the geographical expansion and differentiation of Eurasian populations as they settled the vast expanse of these continents. Consistent with previous proposals14, a parsimonious interpretation of the phylogeny is that the predominant African haplogroup, haplogroup E, arose outside the continent. This model of geographical segregation within the CT clade requires just one continental haplogroup exchange (E to Africa), rather than three (D, C, and F out of Africa). Furthermore, the timing of this putative return to Africa—between the emergence of haplogroup E and its differentiation within Africa by 58 kya—is consistent with proposals, based on non–Y chromosome data, of abundant gene flow between Africa and nearby regions of Asia 50–80 kya15.I've long argued for the Y-chromosome haplogroup E migration into Africa and it is nice to see this common-sense interpretation finally adopted. Too much focus has been placed on figuring out which routes modern humans took out of Africa, and not at all to figure out how Eurasian males came to overwhelm the African Y-chromosome gene pool so decisively. The Eurasian migration into Africa must have taken place in the ~70-60kya window, constrained by the D/E split and the deepest intra-African E splits. I think that the Out-of-Arabia scenario I outlined in 2012 continues to make a lot of sense. It would be awesome to get data from the first Later Stone Age people from Africa which are probably the best bet to trace this migration from Eurasia into Sub-Saharan Africa.

Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences
G David Poznik et al.
We report the sequences of 1,244 human Y chromosomes randomly ascertained from 26 worldwide populations by the 1000 Genomes Project. We discovered more than 65,000 variants, including single-nucleotide variants, multiple-nucleotide variants, insertions and deletions, short tandem repeats, and copy number variants. Of these, copy number variants contribute the greatest predicted functional impact. We constructed a calibrated phylogenetic tree on the basis of binary single-nucleotide variants and projected the more complex variants onto it, estimating the number of mutations for each class. Our phylogeny shows bursts of extreme expansion in male numbers that have occurred independently among each of the five continental superpopulations examined, at times of known migrations and technological innovations.
Link
April 24, 2016
Jewish and Indian ancestry in the Bene Israel
PLoS ONE 11(3): e0152056. doi:10.1371/journal.pone.0152056
The Genetics of Bene Israel from India Reveals Both Substantial Jewish and Indian Ancestry
Yedael Y. Waldman , Arjun Biddanda , Natalie R. Davidson, Paul Billing-Ross, Maya Dubrovsky, Christopher L. Campbell, Carole Oddoux, Eitan Friedman, Gil Atzmon, Eran Halperin, Harry Ostrer, Alon Keinan
The Bene Israel Jewish community from West India is a unique population whose history before the 18th century remains largely unknown. Bene Israel members consider themselves as descendants of Jews, yet the identity of Jewish ancestors and their arrival time to India are unknown, with speculations on arrival time varying between the 8th century BCE and the 6th century CE. Here, we characterize the genetic history of Bene Israel by collecting and genotyping 18 Bene Israel individuals. Combining with 486 individuals from 41 other Jewish, Indian and Pakistani populations, and additional individuals from worldwide populations, we conducted comprehensive genome-wide analyses based on FST, principal component analysis, ADMIXTURE, identity-by-descent sharing, admixture linkage disequilibrium decay, haplotype sharing and allele sharing autocorrelation decay, as well as contrasted patterns between the X chromosome and the autosomes. The genetics of Bene Israel individuals resemble local Indian populations, while at the same time constituting a clearly separated and unique population in India. They are unique among Indian and Pakistani populations we analyzed in sharing considerable genetic ancestry with other Jewish populations. Putting together the results from all analyses point to Bene Israel being an admixed population with both Jewish and Indian ancestry, with the genetic contribution of each of these ancestral populations being substantial. The admixture took place in the last millennium, about 19–33 generations ago. It involved Middle-Eastern Jews and was sex-biased, with more male Jewish and local female contribution. It was followed by a population bottleneck and high endogamy, which can lead to increased prevalence of recessive diseases in this population. This study provides an example of how genetic analysis advances our knowledge of human history in cases where other disciplines lack the relevant data to do so.
Link
The Genetics of Bene Israel from India Reveals Both Substantial Jewish and Indian Ancestry
Yedael Y. Waldman , Arjun Biddanda , Natalie R. Davidson, Paul Billing-Ross, Maya Dubrovsky, Christopher L. Campbell, Carole Oddoux, Eitan Friedman, Gil Atzmon, Eran Halperin, Harry Ostrer, Alon Keinan
The Bene Israel Jewish community from West India is a unique population whose history before the 18th century remains largely unknown. Bene Israel members consider themselves as descendants of Jews, yet the identity of Jewish ancestors and their arrival time to India are unknown, with speculations on arrival time varying between the 8th century BCE and the 6th century CE. Here, we characterize the genetic history of Bene Israel by collecting and genotyping 18 Bene Israel individuals. Combining with 486 individuals from 41 other Jewish, Indian and Pakistani populations, and additional individuals from worldwide populations, we conducted comprehensive genome-wide analyses based on FST, principal component analysis, ADMIXTURE, identity-by-descent sharing, admixture linkage disequilibrium decay, haplotype sharing and allele sharing autocorrelation decay, as well as contrasted patterns between the X chromosome and the autosomes. The genetics of Bene Israel individuals resemble local Indian populations, while at the same time constituting a clearly separated and unique population in India. They are unique among Indian and Pakistani populations we analyzed in sharing considerable genetic ancestry with other Jewish populations. Putting together the results from all analyses point to Bene Israel being an admixed population with both Jewish and Indian ancestry, with the genetic contribution of each of these ancestral populations being substantial. The admixture took place in the last millennium, about 19–33 generations ago. It involved Middle-Eastern Jews and was sex-biased, with more male Jewish and local female contribution. It was followed by a population bottleneck and high endogamy, which can lead to increased prevalence of recessive diseases in this population. This study provides an example of how genetic analysis advances our knowledge of human history in cases where other disciplines lack the relevant data to do so.
Link
April 14, 2016
Periods of human activity in Chauvet-Pont d'Arc cave
PNAS DOI: doi: 10.1073/pnas.1523158113
A high-precision chronological model for the decorated Upper Paleolithic cave of Chauvet-Pont d’Arc, Ardèche, France
Anita Quiles et al.
Radiocarbon dates for the ancient drawings in the Chauvet-Pont d’Arc Cave revealed ages much older than expected. These early ages and nature of this Paleolithic art make this United Nations Educational, Scientific and Cultural Organization (UNESCO) site indisputably unique. A large, multidisciplinary dating program has recently mapped the anthropological evolution associated with the cave. More than 350 dates (by 14C, U-Th, TL and 36Cl) were obtained over the last 15 y. They include 259 radiocarbon dates, mainly related to the rock art and human activity in the cave. We present here more than 80 previously unpublished dates. All of the dates were integrated into a high-precision Bayesian model based on archaeological evidence to securely reconstruct the complete history of the Chauvet-Pont d’Arc Cave on an absolute timescale. It shows that there were two distinct periods of human activity in the cave, one from 37 to 33,500 y ago, and the other from 31 to 28,000 y ago. Cave bears also took refuge in the cave until 33,000 y ago.
Link
A high-precision chronological model for the decorated Upper Paleolithic cave of Chauvet-Pont d’Arc, Ardèche, France
Anita Quiles et al.
Radiocarbon dates for the ancient drawings in the Chauvet-Pont d’Arc Cave revealed ages much older than expected. These early ages and nature of this Paleolithic art make this United Nations Educational, Scientific and Cultural Organization (UNESCO) site indisputably unique. A large, multidisciplinary dating program has recently mapped the anthropological evolution associated with the cave. More than 350 dates (by 14C, U-Th, TL and 36Cl) were obtained over the last 15 y. They include 259 radiocarbon dates, mainly related to the rock art and human activity in the cave. We present here more than 80 previously unpublished dates. All of the dates were integrated into a high-precision Bayesian model based on archaeological evidence to securely reconstruct the complete history of the Chauvet-Pont d’Arc Cave on an absolute timescale. It shows that there were two distinct periods of human activity in the cave, one from 37 to 33,500 y ago, and the other from 31 to 28,000 y ago. Cave bears also took refuge in the cave until 33,000 y ago.
Link
April 10, 2016
Nubian assemblages from the Negev
Nubian assemblages from the Levant are quite important because they provide an intermediate link (along land routes) between those from Africa and Arabia. It's also more difficult now to consider the Arabian finds as a limited event without broader implications about modern human dispersals.
From the paper:
“Diffusion with modifications”: Nubian assemblages in the central Negev highlands of Israel and their implications for Middle Paleolithic inter-regional interactions
Mae Goder-Goldberger, Natalia Gubenko, Erella Hovers
Nubian Levallois cores, now known from sites in eastern Africa, the Nile Valley and Arabia, have been used as a material culture marker for Upper Pleistocene dispersals of hominins out of Africa. The Levantine corridor, being the only land route connecting Africa to Eurasia, has been viewed as a possible dispersal route. We report here on lithic assemblages from the Negev highlands of Israel that contain both Levallois centripetal and Nubian-type cores. Wetter conditions over the Sahara and Negev deserts during MIS 6a–5e provided a generally continuous environmental corridor into the Levant that enabled the dispersal of hominin groups bearing the Nubian variant of prepared core technologies. The Negev assemblages draw renewed attention to the place of the Levant as one of the dispersal routes out of Africa during the Late Pleistocene and could suggest that processes of human dispersals and cultural diffusion resulted in the spread of Nubian technology across eastern Africa, the western Sahara and the Nile Valley, the southern Levant and Arabia.
Link
Mapping the earliest dated sites that contain a Nubian component does not permit an unequivocal identification of a region of origin for the Nubian Technology.Quaternary International doi:10.1016/j.quaint.2016.02.008
“Diffusion with modifications”: Nubian assemblages in the central Negev highlands of Israel and their implications for Middle Paleolithic inter-regional interactions
Mae Goder-Goldberger, Natalia Gubenko, Erella Hovers
Nubian Levallois cores, now known from sites in eastern Africa, the Nile Valley and Arabia, have been used as a material culture marker for Upper Pleistocene dispersals of hominins out of Africa. The Levantine corridor, being the only land route connecting Africa to Eurasia, has been viewed as a possible dispersal route. We report here on lithic assemblages from the Negev highlands of Israel that contain both Levallois centripetal and Nubian-type cores. Wetter conditions over the Sahara and Negev deserts during MIS 6a–5e provided a generally continuous environmental corridor into the Levant that enabled the dispersal of hominin groups bearing the Nubian variant of prepared core technologies. The Negev assemblages draw renewed attention to the place of the Levant as one of the dispersal routes out of Africa during the Late Pleistocene and could suggest that processes of human dispersals and cultural diffusion resulted in the spread of Nubian technology across eastern Africa, the western Sahara and the Nile Valley, the southern Levant and Arabia.
Link
April 07, 2016
Neandertal Y-chromosome (finally)
It's been six years since the publication of the draft Neandertal genome, and one piece of the puzzle that's always been missing is the Neandertal Y-chromosome (unfortunately most of the Neandertals yielding data were female). But, the wait is finally over, with the first publication of Neandertal Y-chromosome data.
AJHG Volume 98, Issue 4, p728–734, 7 April 2016
The Divergence of Neandertal and Modern Human Y Chromosomes
Fernando L. Mendez, G. David Poznik, Sergi Castellano, Carlos D. Bustamante
Sequencing the genomes of extinct hominids has reshaped our understanding of modern human origins. Here, we analyze ∼120 kb of exome-captured Y-chromosome DNA from a Neandertal individual from El Sidrón, Spain. We investigate its divergence from orthologous chimpanzee and modern human sequences and find strong support for a model that places the Neandertal lineage as an outgroup to modern human Y chromosomes—including A00, the highly divergent basal haplogroup. We estimate that the time to the most recent common ancestor (TMRCA) of Neandertal and modern human Y chromosomes is ∼588 thousand years ago (kya) (95% confidence interval [CI]: 447–806 kya). This is ∼2.1 (95% CI: 1.7–2.9) times longer than the TMRCA of A00 and other extant modern human Y-chromosome lineages. This estimate suggests that the Y-chromosome divergence mirrors the population divergence of Neandertals and modern human ancestors, and it refutes alternative scenarios of a relatively recent or super-archaic origin of Neandertal Y chromosomes. The fact that the Neandertal Y we describe has never been observed in modern humans suggests that the lineage is most likely extinct. We identify protein-coding differences between Neandertal and modern human Y chromosomes, including potentially damaging changes to PCDH11Y, TMSB4Y, USP9Y, and KDM5D. Three of these changes are missense mutations in genes that produce male-specific minor histocompatibility (H-Y) antigens. Antigens derived from KDM5D, for example, are thought to elicit a maternal immune response during gestation. It is possible that incompatibilities at one or more of these genes played a role in the reproductive isolation of the two groups.
Link
AJHG Volume 98, Issue 4, p728–734, 7 April 2016
The Divergence of Neandertal and Modern Human Y Chromosomes
Fernando L. Mendez, G. David Poznik, Sergi Castellano, Carlos D. Bustamante
Sequencing the genomes of extinct hominids has reshaped our understanding of modern human origins. Here, we analyze ∼120 kb of exome-captured Y-chromosome DNA from a Neandertal individual from El Sidrón, Spain. We investigate its divergence from orthologous chimpanzee and modern human sequences and find strong support for a model that places the Neandertal lineage as an outgroup to modern human Y chromosomes—including A00, the highly divergent basal haplogroup. We estimate that the time to the most recent common ancestor (TMRCA) of Neandertal and modern human Y chromosomes is ∼588 thousand years ago (kya) (95% confidence interval [CI]: 447–806 kya). This is ∼2.1 (95% CI: 1.7–2.9) times longer than the TMRCA of A00 and other extant modern human Y-chromosome lineages. This estimate suggests that the Y-chromosome divergence mirrors the population divergence of Neandertals and modern human ancestors, and it refutes alternative scenarios of a relatively recent or super-archaic origin of Neandertal Y chromosomes. The fact that the Neandertal Y we describe has never been observed in modern humans suggests that the lineage is most likely extinct. We identify protein-coding differences between Neandertal and modern human Y chromosomes, including potentially damaging changes to PCDH11Y, TMSB4Y, USP9Y, and KDM5D. Three of these changes are missense mutations in genes that produce male-specific minor histocompatibility (H-Y) antigens. Antigens derived from KDM5D, for example, are thought to elicit a maternal immune response during gestation. It is possible that incompatibilities at one or more of these genes played a role in the reproductive isolation of the two groups.
Link
April 01, 2016
Zhiren cave human remains: 116-106 ka old
Quaternary International doi:10.1016/j.quaint.2015.12.088
The age of human remains and associated fauna from Zhiren Cave in Guangxi, southern China
Yanjun Cai et al.
Zhiren Cave in southern China is an important site for the study of the origin and the environmental background of early modern humans. The combination of Elephas kiangnanensis, Elephas maximus, and Megatapirus augustus, indicates an early representative of the typical Asian elephant fauna. Previous U-series dating of flowstone calcite has pinpointed an upper age limit for the fossils of about 100 ka. In order to achieve a better comprehension of the chronology of the modern human and contemporaneous faunal assemblage, paleomagnetic, stratigraphic, and optically stimulated luminescence (OSL) dating methods have been applied to the cave sediments. Paleomagnetic analyses reveal that there is a reversed polarity excursion below the fossiliferous layer. This excursion can be regarded as the Blake excursion event, given the U-series ages of the overlying flowstone calcite, the OSL measurements, the virtual geomagnetic pole (VGP) path of the excursion, the two reverse polarity zones within this excursion event, and the characteristic of the fauna assemblage. The human remains and mammalian fauna assemblage can be bracketed to 116–106 ka. Application of OSL dating leads to erroneous ages, largely due to the uncertainty associated with the estimation on the dose rates.
Link
The age of human remains and associated fauna from Zhiren Cave in Guangxi, southern China
Yanjun Cai et al.
Zhiren Cave in southern China is an important site for the study of the origin and the environmental background of early modern humans. The combination of Elephas kiangnanensis, Elephas maximus, and Megatapirus augustus, indicates an early representative of the typical Asian elephant fauna. Previous U-series dating of flowstone calcite has pinpointed an upper age limit for the fossils of about 100 ka. In order to achieve a better comprehension of the chronology of the modern human and contemporaneous faunal assemblage, paleomagnetic, stratigraphic, and optically stimulated luminescence (OSL) dating methods have been applied to the cave sediments. Paleomagnetic analyses reveal that there is a reversed polarity excursion below the fossiliferous layer. This excursion can be regarded as the Blake excursion event, given the U-series ages of the overlying flowstone calcite, the OSL measurements, the virtual geomagnetic pole (VGP) path of the excursion, the two reverse polarity zones within this excursion event, and the characteristic of the fauna assemblage. The human remains and mammalian fauna assemblage can be bracketed to 116–106 ka. Application of OSL dating leads to erroneous ages, largely due to the uncertainty associated with the estimation on the dose rates.
Link
March 31, 2016
Denisovan ancestry in Oceanians (and some in South Asians)
Current Biology DOI: http://dx.doi.org/10.1016/j.cub.2016.03.037
The Combined Landscape of Denisovan and Neanderthal Ancestry in Present-Day Humans
Sriram Sankararaman et al.
Some present-day humans derive up to ∼5% [ 1 ] of their ancestry from archaic Denisovans, an even larger proportion than the ∼2% from Neanderthals [ 2 ]. We developed methods that can disambiguate the locations of segments of Denisovan and Neanderthal ancestry in present-day humans and applied them to 257 high-coverage genomes from 120 diverse populations, among which were 20 individual Oceanians with high Denisovan ancestry [ 3 ]. In Oceanians, the average size of Denisovan fragments is larger than Neanderthal fragments, implying a more recent average date of Denisovan admixture in the history of these populations (p = 0.00004). We document more Denisovan ancestry in South Asia than is expected based on existing models of history, reflecting a previously undocumented mixture related to archaic humans (p = 0.0013). Denisovan ancestry, just like Neanderthal ancestry, has been deleterious on a modern human genetic background, as reflected by its depletion near genes. Finally, the reduction of both archaic ancestries is especially pronounced on chromosome X and near genes more highly expressed in testes than other tissues (p = 1.2 × 10−7 to 3.2 × 10−7 for Denisovan and 2.2 × 10−3 to 2.9 × 10−3 for Neanderthal ancestry even after controlling for differences in level of selective constraint across gene classes). This suggests that reduced male fertility may be a general feature of mixtures of human populations diverged by >500,000 years.
Link
The Combined Landscape of Denisovan and Neanderthal Ancestry in Present-Day Humans
Sriram Sankararaman et al.
Some present-day humans derive up to ∼5% [ 1 ] of their ancestry from archaic Denisovans, an even larger proportion than the ∼2% from Neanderthals [ 2 ]. We developed methods that can disambiguate the locations of segments of Denisovan and Neanderthal ancestry in present-day humans and applied them to 257 high-coverage genomes from 120 diverse populations, among which were 20 individual Oceanians with high Denisovan ancestry [ 3 ]. In Oceanians, the average size of Denisovan fragments is larger than Neanderthal fragments, implying a more recent average date of Denisovan admixture in the history of these populations (p = 0.00004). We document more Denisovan ancestry in South Asia than is expected based on existing models of history, reflecting a previously undocumented mixture related to archaic humans (p = 0.0013). Denisovan ancestry, just like Neanderthal ancestry, has been deleterious on a modern human genetic background, as reflected by its depletion near genes. Finally, the reduction of both archaic ancestries is especially pronounced on chromosome X and near genes more highly expressed in testes than other tissues (p = 1.2 × 10−7 to 3.2 × 10−7 for Denisovan and 2.2 × 10−3 to 2.9 × 10−3 for Neanderthal ancestry even after controlling for differences in level of selective constraint across gene classes). This suggests that reduced male fertility may be a general feature of mixtures of human populations diverged by >500,000 years.
Link
Middle (not Upper) Paleolithic hobbits
Nature (2016) doi:10.1038/nature17179
Revised stratigraphy and chronology for Homo floresiensis at Liang Bua in Indonesia
Thomas Sutikna, Matthew W. Tocheri, Michael J. Morwood, E. Wahyu Saptomo, Jatmiko, Rokus Due Awe, Sri Wasisto, Kira E. Westaway, Maxime Aubert, Bo Li, Jian-xin Zhao, Michael Storey, Brent V. Alloway, Mike W. Morley, Hanneke J. M. Meijer, Gerrit D. van den Bergh, Rainer Grün, Anthony Dosseto, Adam Brumm, William L. Jungers & Richard G. Roberts
Homo floresiensis, a primitive hominin species discovered in Late Pleistocene sediments at Liang Bua (Flores, Indonesia)1, 2, 3, has generated wide interest and scientific debate. A major reason this taxon is controversial is because the H. floresiensis-bearing deposits, which include associated stone artefacts2, 3, 4 and remains of other extinct endemic fauna5, 6, were dated to between about 95 and 12 thousand calendar years (kyr) ago2, 3, 7. These ages suggested that H. floresiensis survived until long after modern humans reached Australia by ~50 kyr ago8, 9, 10. Here we report new stratigraphic and chronological evidence from Liang Bua that does not support the ages inferred previously for the H. floresiensis holotype (LB1), ~18 thousand calibrated radiocarbon years before present (kyr cal. BP), or the time of last appearance of this species (about 17 or 13–11 kyr cal. BP)1, 2, 3, 7, 11. Instead, the skeletal remains of H. floresiensis and the deposits containing them are dated to between about 100 and 60 kyr ago, whereas stone artefacts attributable to this species range from about 190 to 50 kyr in age. Whether H. floresiensis survived after 50 kyr ago—potentially encountering modern humans on Flores or other hominins dispersing through southeast Asia, such as Denisovans12, 13—is an open question.
Link
Revised stratigraphy and chronology for Homo floresiensis at Liang Bua in Indonesia
Thomas Sutikna, Matthew W. Tocheri, Michael J. Morwood, E. Wahyu Saptomo, Jatmiko, Rokus Due Awe, Sri Wasisto, Kira E. Westaway, Maxime Aubert, Bo Li, Jian-xin Zhao, Michael Storey, Brent V. Alloway, Mike W. Morley, Hanneke J. M. Meijer, Gerrit D. van den Bergh, Rainer Grün, Anthony Dosseto, Adam Brumm, William L. Jungers & Richard G. Roberts
Homo floresiensis, a primitive hominin species discovered in Late Pleistocene sediments at Liang Bua (Flores, Indonesia)1, 2, 3, has generated wide interest and scientific debate. A major reason this taxon is controversial is because the H. floresiensis-bearing deposits, which include associated stone artefacts2, 3, 4 and remains of other extinct endemic fauna5, 6, were dated to between about 95 and 12 thousand calendar years (kyr) ago2, 3, 7. These ages suggested that H. floresiensis survived until long after modern humans reached Australia by ~50 kyr ago8, 9, 10. Here we report new stratigraphic and chronological evidence from Liang Bua that does not support the ages inferred previously for the H. floresiensis holotype (LB1), ~18 thousand calibrated radiocarbon years before present (kyr cal. BP), or the time of last appearance of this species (about 17 or 13–11 kyr cal. BP)1, 2, 3, 7, 11. Instead, the skeletal remains of H. floresiensis and the deposits containing them are dated to between about 100 and 60 kyr ago, whereas stone artefacts attributable to this species range from about 190 to 50 kyr in age. Whether H. floresiensis survived after 50 kyr ago—potentially encountering modern humans on Flores or other hominins dispersing through southeast Asia, such as Denisovans12, 13—is an open question.
Link
March 25, 2016
Bronze Age war in northern Germany
Slaughter at the bridge: Uncovering a colossal Bronze Age battle
About 3200 years ago, two armies clashed at a river crossing near the Baltic Sea. The confrontation can’t be found in any history books—the written word didn’t become common in these parts for another 2000 years—but this was no skirmish between local clans. Thousands of warriors came together in a brutal struggle, perhaps fought on a single day, using weapons crafted from wood, flint, and bronze, a metal that was then the height of military technology.
...
In 1996, an amateur archaeologist found a single upper arm bone sticking out of the steep riverbank—the first clue that the Tollense Valley, about 120 kilometers north of Berlin, concealed a gruesome secret. A flint arrowhead was firmly embedded in one end of the bone, prompting archaeologists to dig a small test excavation that yielded more bones, a bashed-in skull, and a 73-centimeter club resembling a baseball bat. The artifacts all were radiocarbon-dated to about 1250 B.C.E., suggesting they stemmed from a single episode during Europe’s Bronze Age.
...
Northern Europe in the Bronze Age was long dismissed as a backwater, overshadowed by more sophisticated civilizations in the Near East and Greece. Bronze itself, created in the Near East around 3200 B.C.E., took 1000 years to arrive here. But Tollense’s scale suggests more organization—and more violence—than once thought. “We had considered scenarios of raids, with small groups of young men killing and stealing food, but to imagine such a big battle with thousands of people is very surprising,” says Svend Hansen, head of the German Archaeological Institute’s (DAI’s) Eurasia Department in Berlin. The well-preserved bones and artifacts add detail to this picture of Bronze Age sophistication, pointing to the existence of a trained warrior class and suggesting that people from across Europe joined the bloody fray.
...
There was reason for skepticism. Before Tollense, direct evidence of large-scale violence in the Bronze Age was scanty, especially in this region. Historical accounts from the Near East and Greece described epic battles, but few artifacts remained to corroborate these boastful accounts. “Even in Egypt, despite hearing many tales of war, we never find such substantial archaeological evidence of its participants and victims,” UCD’s Molloy says.
...
Ancient DNA could potentially reveal much more: When compared to other Bronze Age samples from around Europe at this time, it could point to the homelands of the warriors as well as such traits as eye and hair color. Genetic analysis is just beginning, but so far it supports the notion of far-flung origins. DNA from teeth suggests some warriors are related to modern southern Europeans and others to people living in modern-day Poland and Scandinavia. “This is not a bunch of local idiots,” says University of Mainz geneticist Joachim Burger. “It’s a highly diverse population.”


March 20, 2016
Neandertal and Denisovan DNA from Melanesians

Admixture models are out of control these days, with 4 inferred archaic introgressions into three groups of Eurasians (Europeans, East Asians, Melanesians). The model on the left has to be a simplification/incomplete/wrong in some way (Melanesians are not an outgroup to Europeans and East Asians; Europeans have "Basal Eurasian" ancestry via Early European Farmers; Denisovans have some kind of weird archaic ancestry that Neandertals don't, and according to a recent study, the Altai Neandertal also has some kind of weird Proto-Modern Human lineage). In any case, this may not matter much for the problem at hand which is excavating archaic DNA from Melanesian genomes.
But, if you combined all the admixtures inferred in the literature, you'd probably need something like 8 admixtures to model 5 populations. Time and data will show which of them are real, and reveal news ones (e.g., in Africans, who remain blissfully simple in the absence of archaic African genomes).
Science DOI: 10.1126/science.aad9416
Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals
Benjamin Vernot et al.
Although Neandertal sequences that persist in the genomes of modern humans have been identified in Eurasians, comparable studies in people whose ancestors hybridized with both Neandertals and Denisovans are lacking. We developed an approach to identify DNA inherited from multiple archaic hominin ancestors and applied it to whole-genome sequences from 1523 geographically diverse individuals, including 35 new Island Melanesian genomes. In aggregate, we recovered 1.34 Gb and 303 Mb of the Neandertal and Denisovan genome, respectively. We leverage these maps of archaic sequence to show that Neandertal admixture occurred multiple times in different non-African populations, characterize genomic regions that are significantly depleted of archaic sequence, and identify signatures of adaptive introgression.
Link
Adaptation in the light of ancient genomes
Nature Communications 7, Article number: 10775 doi:10.1038/ncomms10775
Human adaptation and population differentiation in the light of ancient genomes
Felix M. Key, Qiaomei Fu, Frédéric Romagné, Michael Lachmann and Aida M. Andrés
The influence of positive selection sweeps in human evolution is increasingly debated, although our ability to detect them is hampered by inherent uncertainties in the timing of past events. Ancient genomes provide snapshots of allele frequencies in the past and can help address this question. We combine modern and ancient genomic data in a simple statistic (DAnc) to time allele frequency changes, and investigate the role of drift and adaptation in population differentiation. Only 30% of the most strongly differentiated alleles between Africans and Eurasians changed in frequency during the colonization of Eurasia, but in Europe these alleles are enriched in genic and putatively functional alleles to an extent only compatible with local adaptation. Adaptive alleles—especially those associated with pigmentation—are mostly of hunter-gatherer origin, although lactose persistence arose in a haplotype present in farmers. These results provide evidence for a role of local adaptation in human population differentiation.
Link
Human adaptation and population differentiation in the light of ancient genomes
Felix M. Key, Qiaomei Fu, Frédéric Romagné, Michael Lachmann and Aida M. Andrés
The influence of positive selection sweeps in human evolution is increasingly debated, although our ability to detect them is hampered by inherent uncertainties in the timing of past events. Ancient genomes provide snapshots of allele frequencies in the past and can help address this question. We combine modern and ancient genomic data in a simple statistic (DAnc) to time allele frequency changes, and investigate the role of drift and adaptation in population differentiation. Only 30% of the most strongly differentiated alleles between Africans and Eurasians changed in frequency during the colonization of Eurasia, but in Europe these alleles are enriched in genic and putatively functional alleles to an extent only compatible with local adaptation. Adaptive alleles—especially those associated with pigmentation—are mostly of hunter-gatherer origin, although lactose persistence arose in a haplotype present in farmers. These results provide evidence for a role of local adaptation in human population differentiation.
Link
March 15, 2016
Preprint revolution in biology
A very nice article by Amy Harmon in the NY Times.
Handful of Biologists Went Rogue and Published Directly to Internet
Handful of Biologists Went Rogue and Published Directly to Internet
On Feb. 29, Carol Greider of Johns Hopkins University became the third Nobel Prize laureate biologist in a month to do something long considered taboo among biomedical researchers: She posted a report of her recent discoveries to a publicly accessible website, bioRxiv, before submitting it to a scholarly journal to review for “official’’ publication.
...
And many #ASAPbio supporters retweeted John Hawks, a paleoanthropologist from the University of Wisconsin, who found himself recently at an African university where a paper on African genomes was unavailable because it could not pay the fee for the journal where it was published, and no preprint was available. He expressed his frustration with a profanity.
...
Preprint advocates counter that scientists care too much about their reputations to publish shoddy work, and posts to bioRxiv are clearly marked to indicate that they may contain information that “has not yet been accepted or endorsed in any way by the scientific or medical community.’’ Others note that plenty of peer-reviewed papers in high-profile journals have proved to be wrong, and some argue that carrying out peer review after a paper is published would provide a more rigorous and fair vetting of papers, anyway.
March 14, 2016
The dysgenic effects of modern civilization
I hope that strong AI and practical gene editing becomes a reality before this bleak future kicks in.
GENETICS March 7, 2016 vol. 202 no. 3 869-875; DOI: 10.1534/genetics.115.180471
Mutation and Human Exceptionalism: Our Future Genetic Load
Michael Lynch
Although the human germline mutation rate is higher than that in any other well-studied species, the rate is not exceptional once the effective genome size and effective population size are taken into consideration. Human somatic mutation rates are substantially elevated above those in the germline, but this is also seen in other species. What is exceptional about humans is the recent detachment from the challenges of the natural environment and the ability to modify phenotypic traits in ways that mitigate the fitness effects of mutations, e.g., precision and personalized medicine. This results in a relaxation of selection against mildly deleterious mutations, including those magnifying the mutation rate itself. The long-term consequence of such effects is an expected genetic deterioration in the baseline human condition, potentially measurable on the timescale of a few generations in westernized societies, and because the brain is a particularly large mutational target, this is of particular concern. Ultimately, the price will have to be covered by further investment in various forms of medical intervention. Resolving the uncertainties of the magnitude and timescale of these effects will require the establishment of stable, standardized, multigenerational measurement procedures for various human traits.
Link
GENETICS March 7, 2016 vol. 202 no. 3 869-875; DOI: 10.1534/genetics.115.180471
Mutation and Human Exceptionalism: Our Future Genetic Load
Michael Lynch
Although the human germline mutation rate is higher than that in any other well-studied species, the rate is not exceptional once the effective genome size and effective population size are taken into consideration. Human somatic mutation rates are substantially elevated above those in the germline, but this is also seen in other species. What is exceptional about humans is the recent detachment from the challenges of the natural environment and the ability to modify phenotypic traits in ways that mitigate the fitness effects of mutations, e.g., precision and personalized medicine. This results in a relaxation of selection against mildly deleterious mutations, including those magnifying the mutation rate itself. The long-term consequence of such effects is an expected genetic deterioration in the baseline human condition, potentially measurable on the timescale of a few generations in westernized societies, and because the brain is a particularly large mutational target, this is of particular concern. Ultimately, the price will have to be covered by further investment in various forms of medical intervention. Resolving the uncertainties of the magnitude and timescale of these effects will require the establishment of stable, standardized, multigenerational measurement procedures for various human traits.
Link
Sima de los Huesos hominins were Proto-Neandertals
Nature (2016) doi:10.1038/nature17405
Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins
Matthias Meyer, Juan-Luis Arsuaga, Cesare de Filippo, Sarah Nagel, Ayinuer Aximu-Petri, Birgit Nickel, Ignacio Martínez, Ana Gracia, José María Bermúdez de Castro, Eudald Carbonell, Bence Viola, Janet Kelso, Kay Prüfer & Svante Pääbo
A unique assemblage of 28 hominin individuals, found in Sima de los Huesos in the Sierra de Atapuerca in Spain, has recently been dated to approximately 430,000 years ago1. An interesting question is how these Middle Pleistocene hominins were related to those who lived in the Late Pleistocene epoch, in particular to Neanderthals in western Eurasia and to Denisovans, a sister group of Neanderthals so far known only from southern Siberia. While the Sima de los Huesos hominins share some derived morphological features with Neanderthals, the mitochondrial genome retrieved from one individual from Sima de los Huesos is more closely related to the mitochondrial DNA of Denisovans than to that of Neanderthals2. However, since the mitochondrial DNA does not reveal the full picture of relationships among populations, we have investigated DNA preservation in several individuals found at Sima de los Huesos. Here we recover nuclear DNA sequences from two specimens, which show that the Sima de los Huesos hominins were related to Neanderthals rather than to Denisovans, indicating that the population divergence between Neanderthals and Denisovans predates 430,000 years ago. A mitochondrial DNA recovered from one of the specimens shares the previously described relationship to Denisovan mitochondrial DNAs, suggesting, among other possibilities, that the mitochondrial DNA gene pool of Neanderthals turned over later in their history.
Link
Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins
Matthias Meyer, Juan-Luis Arsuaga, Cesare de Filippo, Sarah Nagel, Ayinuer Aximu-Petri, Birgit Nickel, Ignacio Martínez, Ana Gracia, José María Bermúdez de Castro, Eudald Carbonell, Bence Viola, Janet Kelso, Kay Prüfer & Svante Pääbo
A unique assemblage of 28 hominin individuals, found in Sima de los Huesos in the Sierra de Atapuerca in Spain, has recently been dated to approximately 430,000 years ago1. An interesting question is how these Middle Pleistocene hominins were related to those who lived in the Late Pleistocene epoch, in particular to Neanderthals in western Eurasia and to Denisovans, a sister group of Neanderthals so far known only from southern Siberia. While the Sima de los Huesos hominins share some derived morphological features with Neanderthals, the mitochondrial genome retrieved from one individual from Sima de los Huesos is more closely related to the mitochondrial DNA of Denisovans than to that of Neanderthals2. However, since the mitochondrial DNA does not reveal the full picture of relationships among populations, we have investigated DNA preservation in several individuals found at Sima de los Huesos. Here we recover nuclear DNA sequences from two specimens, which show that the Sima de los Huesos hominins were related to Neanderthals rather than to Denisovans, indicating that the population divergence between Neanderthals and Denisovans predates 430,000 years ago. A mitochondrial DNA recovered from one of the specimens shares the previously described relationship to Denisovan mitochondrial DNAs, suggesting, among other possibilities, that the mitochondrial DNA gene pool of Neanderthals turned over later in their history.
Link
March 09, 2016
Stature/body mass index and socioeconomic status
BMJ 2016; 352 doi: http://dx.doi.org/10.1136/bmj.i582 (Published 08 March 2016)
Height, body mass index, and socioeconomic status: mendelian randomisation study in UK Biobank
Jessica Tyrrell, research fellow1 2, Samuel E Jones, associate research fellow1, Robin Beaumont, associate research fellow1, Christina M Astley, research fellow3 4, Rebecca Lovell, research fellow2, Hanieh Yaghootkar, research fellow1, Marcus Tuke, associate research fellow1, Katherine S Ruth, associate research fellow1, Rachel M Freathy, senior research fellow1, Joel N Hirschhorn, professor2 3 5, Andrew R Wood, research fellow1, Anna Murray, senior lecturer1, Michael N Weedon, associate professor1, Timothy M Frayling, professor1
Abstract
Objective To determine whether height and body mass index (BMI) have a causal role in five measures of socioeconomic status.
Design Mendelian randomisation study to test for causal effects of differences in stature and BMI on five measures of socioeconomic status. Mendelian randomisation exploits the fact that genotypes are randomly assigned at conception and thus not confounded by non-genetic factors.
Setting UK Biobank.
Participants 119 669 men and women of British ancestry, aged between 37 and 73 years.
Main outcome measures Age completed full time education, degree level education, job class, annual household income, and Townsend deprivation index.
Results In the UK Biobank study, shorter stature and higher BMI were observationally associated with several measures of lower socioeconomic status. The associations between shorter stature and lower socioeconomic status tended to be stronger in men, and the associations between higher BMI and lower socioeconomic status tended to be stronger in women. For example, a 1 standard deviation (SD) higher BMI was associated with a £210 (€276; $300; 95% confidence interval £84 to £420; P=6×10−3) lower annual household income in men and a £1890 (£1680 to £2100; P=6×10−15) lower annual household income in women. Genetic analysis provided evidence that these associations were partly causal. A genetically determined 1 SD (6.3 cm) taller stature caused a 0.06 (0.02 to 0.09) year older age of completing full time education (P=0.01), a 1.12 (1.07 to 1.18) times higher odds of working in a skilled profession (P=6×10−7), and a £1130 (£680 to £1580) higher annual household income (P=4×10−8). Associations were stronger in men. A genetically determined 1 SD higher BMI (4.6 kg/m2) caused a £2940 (£1680 to £4200; P=1×10−5) lower annual household income and a 0.10 (0.04 to 0.16) SD (P=0.001) higher level of deprivation in women only.
Conclusions These data support evidence that height and BMI play an important partial role in determining several aspects of a person’s socioeconomic status, especially women’s BMI for income and deprivation and men’s height for education, income, and job class. These findings have important social and health implications, supporting evidence that overweight people, especially women, are at a disadvantage and that taller people, especially men, are at an advantage.
Link
Height, body mass index, and socioeconomic status: mendelian randomisation study in UK Biobank
Jessica Tyrrell, research fellow1 2, Samuel E Jones, associate research fellow1, Robin Beaumont, associate research fellow1, Christina M Astley, research fellow3 4, Rebecca Lovell, research fellow2, Hanieh Yaghootkar, research fellow1, Marcus Tuke, associate research fellow1, Katherine S Ruth, associate research fellow1, Rachel M Freathy, senior research fellow1, Joel N Hirschhorn, professor2 3 5, Andrew R Wood, research fellow1, Anna Murray, senior lecturer1, Michael N Weedon, associate professor1, Timothy M Frayling, professor1
Abstract
Objective To determine whether height and body mass index (BMI) have a causal role in five measures of socioeconomic status.
Design Mendelian randomisation study to test for causal effects of differences in stature and BMI on five measures of socioeconomic status. Mendelian randomisation exploits the fact that genotypes are randomly assigned at conception and thus not confounded by non-genetic factors.
Setting UK Biobank.
Participants 119 669 men and women of British ancestry, aged between 37 and 73 years.
Main outcome measures Age completed full time education, degree level education, job class, annual household income, and Townsend deprivation index.
Results In the UK Biobank study, shorter stature and higher BMI were observationally associated with several measures of lower socioeconomic status. The associations between shorter stature and lower socioeconomic status tended to be stronger in men, and the associations between higher BMI and lower socioeconomic status tended to be stronger in women. For example, a 1 standard deviation (SD) higher BMI was associated with a £210 (€276; $300; 95% confidence interval £84 to £420; P=6×10−3) lower annual household income in men and a £1890 (£1680 to £2100; P=6×10−15) lower annual household income in women. Genetic analysis provided evidence that these associations were partly causal. A genetically determined 1 SD (6.3 cm) taller stature caused a 0.06 (0.02 to 0.09) year older age of completing full time education (P=0.01), a 1.12 (1.07 to 1.18) times higher odds of working in a skilled profession (P=6×10−7), and a £1130 (£680 to £1580) higher annual household income (P=4×10−8). Associations were stronger in men. A genetically determined 1 SD higher BMI (4.6 kg/m2) caused a £2940 (£1680 to £4200; P=1×10−5) lower annual household income and a 0.10 (0.04 to 0.16) SD (P=0.001) higher level of deprivation in women only.
Conclusions These data support evidence that height and BMI play an important partial role in determining several aspects of a person’s socioeconomic status, especially women’s BMI for income and deprivation and men’s height for education, income, and job class. These findings have important social and health implications, supporting evidence that overweight people, especially women, are at a disadvantage and that taller people, especially men, are at an advantage.
Link
February 26, 2016
No Y-chromosomes of recent Indian origin in Australians
Current Biology http://dx.doi.org/10.1016/j.cub.2016.01.028
Deep Roots for Aboriginal Australian Y Chromosomes
Anders Bergström et al.
Australia was one of the earliest regions outside Africa to be colonized by fully modern humans, with archaeological evidence for human presence by 47,000 years ago (47 kya) widely accepted [ 1, 2 ]. However, the extent of subsequent human entry before the European colonial age is less clear. The dingo reached Australia about 4 kya, indirectly implying human contact, which some have linked to changes in language and stone tool technology to suggest substantial cultural changes at the same time [ 3 ]. Genetic data of two kinds have been proposed to support gene flow from the Indian subcontinent to Australia at this time, as well: first, signs of South Asian admixture in Aboriginal Australian genomes have been reported on the basis of genome-wide SNP data [ 4 ]; and second, a Y chromosome lineage designated haplogroup C∗, present in both India and Australia, was estimated to have a most recent common ancestor around 5 kya and to have entered Australia from India [ 5 ]. Here, we sequence 13 Aboriginal Australian Y chromosomes to re-investigate their divergence times from Y chromosomes in other continents, including a comparison of Aboriginal Australian and South Asian haplogroup C chromosomes. We find divergence times dating back to ∼50 kya, thus excluding the Y chromosome as providing evidence for recent gene flow from India into Australia.
Link
Deep Roots for Aboriginal Australian Y Chromosomes
Anders Bergström et al.
Australia was one of the earliest regions outside Africa to be colonized by fully modern humans, with archaeological evidence for human presence by 47,000 years ago (47 kya) widely accepted [ 1, 2 ]. However, the extent of subsequent human entry before the European colonial age is less clear. The dingo reached Australia about 4 kya, indirectly implying human contact, which some have linked to changes in language and stone tool technology to suggest substantial cultural changes at the same time [ 3 ]. Genetic data of two kinds have been proposed to support gene flow from the Indian subcontinent to Australia at this time, as well: first, signs of South Asian admixture in Aboriginal Australian genomes have been reported on the basis of genome-wide SNP data [ 4 ]; and second, a Y chromosome lineage designated haplogroup C∗, present in both India and Australia, was estimated to have a most recent common ancestor around 5 kya and to have entered Australia from India [ 5 ]. Here, we sequence 13 Aboriginal Australian Y chromosomes to re-investigate their divergence times from Y chromosomes in other continents, including a comparison of Aboriginal Australian and South Asian haplogroup C chromosomes. We find divergence times dating back to ∼50 kya, thus excluding the Y chromosome as providing evidence for recent gene flow from India into Australia.
Link
February 20, 2016
Are living Africans nested within Eurasian genetic variation (?)

(Early modern human lineage detected as admixture in the Altai Neandertal, ((Asians, Europeans), Africans)),
and then there are two deeper layers of Eurasian hominins (Neandertal/Denisovans) and the "Mystery hominin" that mixed into Denisovans.
Africans are thus just a leaf of the Eurasian family tree, casting serious doubt -if this model is to be believed- to the position that H. sapiens originated in Africa and are descended from people who never left the continent. It seems much simpler to derive them from an early migration (~200kya?) from Asia which would nicely explain why the continent's first sapiens populations appear tentatively in the northeastern corner, and why they do not replace archaic hominins for most of the 200 thousand years until today. In a reversal of perspective it is not Skhul/Qafzeh that are the "migration that failed", but rather the Omo 1 outlier is.
One might argue that this is just a consequence of the fact that lots of ancient genomes have been published from Eurasia, but none from Africa. So, there are all these branches of deep archaic Eurasians simply because there are no genomes of deep archaic Africans.
But, this explanation does not really work. If Africans had any significant ancestry deeper than the split of "Early modern human lineage", then this lineage would be closer to (Asians, Europeans) than to Africans. However, Kulhwilm et al. assert that it is "equally related to present-day Africans and non-Africans". If they had any ancestry deeper than ((Denisovans, Neandertals), H. sapiens), then (Denisovans, Neandertals) would be closer to non-Africans than to Africans. Well, they are, but this is now satisfactorily explained by admixture from (Denisovans, Neandertals) into non-Africans, thanks to genomes like Ust Ishim, K14, and Oase which have big chunks of Neandertal ancestry that can't be explained any other way. No need to invoke any such lineage when a simpler well-documented alternative exists.
The presented phylogeny negates the possibility of the existence of collateral archaic African kin of the extant Africans that admixed with them, and leads to the conclusion that Africans are nested within Eurasian variation because they really are. This is, of course, incompatible with the statistically inferred archaic introgression into Africans which indeed postulates the existence of such archaic Africans and their contribution to extant ones.
I don't see any obvious flaw with Kulhwilm et al. but if its model is right, then it does lead to some rather extreme conclusions. It contradicts the evidence for archaic introgression; if Hsieh et al. is wrong (and I don't seen any evidence for that either), then Kulhwilm et al. can be saved, but only if Africans are really nested within several layers of Eurasian variation and did not admix at all with the morphologically diverse archaic Africans of the paleoanthropological record. This also doesn't seem right now that we know that sapiens-archaic admixture was a common occurrence in Eurasia. The reversal of perspective alluded to above may help here by removing the opportunity for admixture, but that too is, of course, an extraordinary claim.
In sum, I am rather convinced that the latest discoveries have muddled the origin story of our species and some major rethink is needed to evaluate the totality of the evidence.
February 19, 2016
Archaic introgression in Pygmies
We must remember that detecting archaic admixture in Africa is a statistical power game where only a particular type of such introgression can be detected:
First, it needs to be from highly diverged Palaeoafrican sources so that it will look very different from plain H. sapiens DNA. Unlike Eurasia, there's no genome of an ancient Palaeoafrican one can compare against. All inference is based on African genomes having an improbable amount of heterozygosity in parts of their genome.
Second, it needs to have happened recently enough so that it will come in big chunks that can be distinguished from the plain H. sapiens background. Given enough time, recombination breaks down archaic segments into ever tinier bits. You can argue that an unusually long divergent haplotype with a deep TMRCA is archaic, but you can't argue that a single SNP is.
I have little doubt that most if not all of the supposedly "old divergences" between African populations are a mirage created by admixture between modern humans and archaic "Palaeoafricans" diverging and admixing at different time depths. The palaeoanthropological record is quite clear that modern humans were not the only game in town for most of the 200 thousand years since modern humans first appeared in the continent's northeastern corner.
A handful or two of archaic genomes from Eurasia needs an ever-more-complex web of admixtures to make sense of; Africa will need no less, and -if morphological variability persistence is any criterion- a lot more.
Genome Research Published in Advance February 17, 2016, doi: 10.1101/gr.196634.115
Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies
PingHsun Hsieh et al.
Comparisons of whole-genome sequences from ancient and contemporary samples have pointed to several instances of archaic admixture through interbreeding between the ancestors of modern non-Africans and now extinct hominids such as Neanderthals and Denisovans. One implication of these findings is that some adaptive features in contemporary humans may have entered the population via gene flow with archaic forms in Eurasia. Within Africa, fossil evidence suggests that anatomically modern humans (AMH) and various archaic forms coexisted for much of the last 200,000 yr; however, the absence of ancient DNA in Africa has limited our ability to make a direct comparison between archaic and modern human genomes. Here, we use statistical inference based on high coverage whole-genome data (greater than 60×) from contemporary African Pygmy hunter-gatherers as an alternative means to study the evolutionary history of the genus Homo. Using whole-genome simulations that consider demographic histories that include both isolation and gene flow with neighboring farming populations, our inference method rejects the hypothesis that the ancestors of AMH were genetically isolated in Africa, thus providing the first whole genome-level evidence of African archaic admixture. Our inferences also suggest a complex human evolutionary history in Africa, which involves at least a single admixture event from an unknown archaic population into the ancestors of AMH, likely within the last 30,000 yr.
Link
Genome Research Published in Advance February 17, 2016, doi: 10.1101/gr.192971.115
Whole-genome sequence analyses of Western Central African Pygmy hunter-gatherers reveal a complex demographic history and identify candidate genes under positive natural selection
PingHsun Hsieh et al.
African Pygmies practicing a mobile hunter-gatherer lifestyle are phenotypically and genetically diverged from other anatomically modern humans, and they likely experienced strong selective pressures due to their unique lifestyle in the Central African rainforest. To identify genomic targets of adaptation, we sequenced the genomes of four Biaka Pygmies from the Central African Republic and jointly analyzed these data with the genome sequences of three Baka Pygmies from Cameroon and nine Yoruba famers. To account for the complex demographic history of these populations that includes both isolation and gene flow, we fit models using the joint allele frequency spectrum and validated them using independent approaches. Our two best-fit models both suggest ancient divergence between the ancestors of the farmers and Pygmies, 90,000 or 150,000 yr ago. We also find that bidirectional asymmetric gene flow is statistically better supported than a single pulse of unidirectional gene flow from farmers to Pygmies, as previously suggested. We then applied complementary statistics to scan the genome for evidence of selective sweeps and polygenic selection. We found that conventional statistical outlier approaches were biased toward identifying candidates in regions of high mutation or low recombination rate. To avoid this bias, we assigned P-values for candidates using whole-genome simulations incorporating demography and variation in both recombination and mutation rates. We found that genes and gene sets involved in muscle development, bone synthesis, immunity, reproduction, cell signaling and development, and energy metabolism are likely to be targets of positive natural selection in Western African Pygmies or their recent ancestors.
Link
First, it needs to be from highly diverged Palaeoafrican sources so that it will look very different from plain H. sapiens DNA. Unlike Eurasia, there's no genome of an ancient Palaeoafrican one can compare against. All inference is based on African genomes having an improbable amount of heterozygosity in parts of their genome.
Second, it needs to have happened recently enough so that it will come in big chunks that can be distinguished from the plain H. sapiens background. Given enough time, recombination breaks down archaic segments into ever tinier bits. You can argue that an unusually long divergent haplotype with a deep TMRCA is archaic, but you can't argue that a single SNP is.
I have little doubt that most if not all of the supposedly "old divergences" between African populations are a mirage created by admixture between modern humans and archaic "Palaeoafricans" diverging and admixing at different time depths. The palaeoanthropological record is quite clear that modern humans were not the only game in town for most of the 200 thousand years since modern humans first appeared in the continent's northeastern corner.
A handful or two of archaic genomes from Eurasia needs an ever-more-complex web of admixtures to make sense of; Africa will need no less, and -if morphological variability persistence is any criterion- a lot more.
Genome Research Published in Advance February 17, 2016, doi: 10.1101/gr.196634.115
Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies
PingHsun Hsieh et al.
Comparisons of whole-genome sequences from ancient and contemporary samples have pointed to several instances of archaic admixture through interbreeding between the ancestors of modern non-Africans and now extinct hominids such as Neanderthals and Denisovans. One implication of these findings is that some adaptive features in contemporary humans may have entered the population via gene flow with archaic forms in Eurasia. Within Africa, fossil evidence suggests that anatomically modern humans (AMH) and various archaic forms coexisted for much of the last 200,000 yr; however, the absence of ancient DNA in Africa has limited our ability to make a direct comparison between archaic and modern human genomes. Here, we use statistical inference based on high coverage whole-genome data (greater than 60×) from contemporary African Pygmy hunter-gatherers as an alternative means to study the evolutionary history of the genus Homo. Using whole-genome simulations that consider demographic histories that include both isolation and gene flow with neighboring farming populations, our inference method rejects the hypothesis that the ancestors of AMH were genetically isolated in Africa, thus providing the first whole genome-level evidence of African archaic admixture. Our inferences also suggest a complex human evolutionary history in Africa, which involves at least a single admixture event from an unknown archaic population into the ancestors of AMH, likely within the last 30,000 yr.
Link
Genome Research Published in Advance February 17, 2016, doi: 10.1101/gr.192971.115
Whole-genome sequence analyses of Western Central African Pygmy hunter-gatherers reveal a complex demographic history and identify candidate genes under positive natural selection
PingHsun Hsieh et al.
African Pygmies practicing a mobile hunter-gatherer lifestyle are phenotypically and genetically diverged from other anatomically modern humans, and they likely experienced strong selective pressures due to their unique lifestyle in the Central African rainforest. To identify genomic targets of adaptation, we sequenced the genomes of four Biaka Pygmies from the Central African Republic and jointly analyzed these data with the genome sequences of three Baka Pygmies from Cameroon and nine Yoruba famers. To account for the complex demographic history of these populations that includes both isolation and gene flow, we fit models using the joint allele frequency spectrum and validated them using independent approaches. Our two best-fit models both suggest ancient divergence between the ancestors of the farmers and Pygmies, 90,000 or 150,000 yr ago. We also find that bidirectional asymmetric gene flow is statistically better supported than a single pulse of unidirectional gene flow from farmers to Pygmies, as previously suggested. We then applied complementary statistics to scan the genome for evidence of selective sweeps and polygenic selection. We found that conventional statistical outlier approaches were biased toward identifying candidates in regions of high mutation or low recombination rate. To avoid this bias, we assigned P-values for candidates using whole-genome simulations incorporating demography and variation in both recombination and mutation rates. We found that genes and gene sets involved in muscle development, bone synthesis, immunity, reproduction, cell signaling and development, and energy metabolism are likely to be targets of positive natural selection in Western African Pygmies or their recent ancestors.
Link