AJHG has a preprint of an article about admixture mapping in African Americans. It seems that the next-generation SNP tests for admixture estimation, using thousands, as opposed to hundreds of markers are already here. It is probably a matter of time until someone will be able to create tests to determine "national origin", even though in many cases where nations have been formed by recent linguistic/religious/political differences such tests may prove to be impossible.
American Journal of Human Genetics (preprint)
A Genome-Wide SNP panel with High Ancestry Information for African American Admixture
Mapping
Chao Tian et al.
Abstract
Admixture mapping requires a genome-wide panel of relatively evenly spaced markers that can distinguish the ancestral origins of chromosomal segments in admixed individuals. Using the results of the International HapMap Project and specific selection criteria the current study has
examined the ability of selected SNPs to extract continental ancestry information in African American subjects and to explore parameters for admixture mapping. Genotyping of two linguistically diverse west African populations (Bini and Kanuri Nigerians who are Niger-Congo
(Bantu) and Nilo-Saharan speakers, respectively), European Americans and African Americans validated a genome-wide set of >4000 SNP ancestry informative markers with mean and median Fst values > 0.59 and mean and median Fisher’s information content >2.5. This set of SNPs extracted a larger amount of ancestry information in African Americans than previous reported SNP panels and provides nearly uniform coverage of the genome. Moreover in the current study, simulations show that this more informative panel improves power for admixture mapping in
African Americans when ethnicity risk ratios are modest. This is particularly important in the application of admixture mapping in complex genetic diseases for which only modest ethnicity risk ratios of relevant susceptibility genes are expected.
Link

July 27, 2006
mtDNA of Australians
Intern Med J. 2006 Aug;36(8):530-3.
Prevalence of mitochondrial DNA haplogroups in an Australian population.
Manwaring N, Jones MM, Wang JJ, Rochtchina E, Mitchell P, Sue CM.
Mitochondrial DNA (mtDNA) haplogroups are 'neutral polymorphisms' in the mtDNA genome, which have accumulated and persisted along maternal lineages as the human population has migrated worldwide. Three ethnically distinct lineages of human mtDNA populations have been identified: European, characterized by nine haplogroups H, I, J, K, T, U, V, W and X; African, characterized by superhaplogroup L and Asian, characterized by superhaplogroup M. We studied the prevalence of mtDNA haplogroups in participants of the Blue Mountains Eye Study, a large population-based survey of vision conducted between 1991 and 2000 of non-institutionalized permanent residents aged 49 years or older from two suburban postcode areas, west of Sydney, Australia. Total DNA isolated from either hair follicles or blood was available for 3377 of the 3509 participants (96.2%) to determine mtDNA haplogroups by polymerase chain reaction/restriction fragment length polymorphism analysis. Approximately 94.2% of samples could be assigned to one of the nine major European haplogroups, whereas a further 1.2% included the African (L) and Asian (M) superhaplogroups. The five principal haplogroups represented were H (42.9%), U (14.1%), J (10.7%), T (9.2%) and K (8.1%), which together included 85% of this population.
Link
Prevalence of mitochondrial DNA haplogroups in an Australian population.
Manwaring N, Jones MM, Wang JJ, Rochtchina E, Mitchell P, Sue CM.
Mitochondrial DNA (mtDNA) haplogroups are 'neutral polymorphisms' in the mtDNA genome, which have accumulated and persisted along maternal lineages as the human population has migrated worldwide. Three ethnically distinct lineages of human mtDNA populations have been identified: European, characterized by nine haplogroups H, I, J, K, T, U, V, W and X; African, characterized by superhaplogroup L and Asian, characterized by superhaplogroup M. We studied the prevalence of mtDNA haplogroups in participants of the Blue Mountains Eye Study, a large population-based survey of vision conducted between 1991 and 2000 of non-institutionalized permanent residents aged 49 years or older from two suburban postcode areas, west of Sydney, Australia. Total DNA isolated from either hair follicles or blood was available for 3377 of the 3509 participants (96.2%) to determine mtDNA haplogroups by polymerase chain reaction/restriction fragment length polymorphism analysis. Approximately 94.2% of samples could be assigned to one of the nine major European haplogroups, whereas a further 1.2% included the African (L) and Asian (M) superhaplogroups. The five principal haplogroups represented were H (42.9%), U (14.1%), J (10.7%), T (9.2%) and K (8.1%), which together included 85% of this population.
Link
European population substructure revealed by genetics
The free journal PLoS Genetics has a new article which shows that Mediterranean Europeans can be fairly well distinguished from other Europeans using a genome-wide panel comprising 5,700+ SNP markers.
PLoS Genetics (early online release)
European Population Substructure: Clustering of Northern and Southern Populations
Michael F Seldin et al.
PROVISIONAL ABSTRACT
The development of methodologies for defining population genetic structure has provided the ability to identify the major ethnic contributions in individual subjects in diverse populations. Using a genome-wide SNP panel we observe population structure in a diverse group of Europeans and European Americans. Under a variety of conditions and tests there is a consistent and reproducible distinction between "northern" and "southern" European population groups: most individual subjects with southern European ancestry (Italian, Spanish, Portuguese, and Greek) have >85% membership in the "south" population; and most northern, western, eastern and central Europeans have >90% in the "north" population group. Ashkenazi Jewish as well as Sephardic Jewish origin also showed >85% membership in the "south" population consistent with a later Mediterranean origin of these ethnic groups. Based on this work, we have developed a core set of informative SNP markers that can control for this partition in European population structure in a variety of clinical and genetic studies.
Link
PLoS Genetics (early online release)
European Population Substructure: Clustering of Northern and Southern Populations
Michael F Seldin et al.
PROVISIONAL ABSTRACT
The development of methodologies for defining population genetic structure has provided the ability to identify the major ethnic contributions in individual subjects in diverse populations. Using a genome-wide SNP panel we observe population structure in a diverse group of Europeans and European Americans. Under a variety of conditions and tests there is a consistent and reproducible distinction between "northern" and "southern" European population groups: most individual subjects with southern European ancestry (Italian, Spanish, Portuguese, and Greek) have >85% membership in the "south" population; and most northern, western, eastern and central Europeans have >90% in the "north" population group. Ashkenazi Jewish as well as Sephardic Jewish origin also showed >85% membership in the "south" population consistent with a later Mediterranean origin of these ethnic groups. Based on this work, we have developed a core set of informative SNP markers that can control for this partition in European population structure in a variety of clinical and genetic studies.
Link
Black American marriage patterns
Interesting press release showing how native-born American blacks differ from foreign-born ones in their tendency to marry outside their group.
Native-born blacks more likely to marry whites than other blacks
Native-born blacks more likely to marry whites than other blacks
Breaking away from previous marriage and cohabitation studies that treated the U.S. black population as a monolithic culture, a new Cornell study finds significant variations in interracial marriage statistics among American-born blacks and black immigrants from the Caribbean and Africa.
Among the findings: Blacks born on the U.S. mainland (African-Americans) are more likely to cohabit with and marry whites than are newcomers from the West Indies, Africa and Puerto Rico, who are more likely to marry within their group or to marry African-Americans, although marriage rates between these two groups remain very low.
The study also found that there is a higher proportion of cohabitation among interracial couples relative to mixed race marriages, said Daniel T. Lichter, professor of policy analysis and management and director of the Bronfenbrenner Life Course Center at Cornell. "Nearly 10 percent of all cohabiting unions are between partners of different races."
The study, conducted with Christie Batson and Zhenchao Qian of Ohio State University, is published in the August issue of the Journal of Marriage and Family.
Among the study's other findings:
* A higher percentage of black cohabiting couples than married couples involve partners of different races or ethnic ancestries;
* Nearly 75 percent of interracial couples in which one partner is black involve a black man and white woman;
* African-Americans are more likely to pair with blacks born outside the United States than with whites.
The study is an effort to tease out discrete factors that influence "assimilation" -- a term often interpreted as pejorative -- of minority groups into the majority population. The recent increase in interracial unions presumably reflects positive changes in American race relations and a blurring of racial and ethnic boundaries and identities, Lichter said.
However, relying on data from the 2000 census, the primary goal of the study, he said, "is to acknowledge the diversity in America's black populations, while highlighting emerging patterns of marriage and cohabitation with whites as well as other black subpopulations."
Demographers and sociologists have always treated intermarriage between groups -- blacks and whites, or between whites of different ethnic backgrounds -- as evidence of a narrowing of "social distance" between groups, Lichter said. Quantifying these relationships is a useful way to summarize the extent to which minority or historically oppressed populations -- including new immigrants -- are being economically, geographically and politically incorporated into American society and to which racial boundaries are breaking down.
"A current question is whether the new black population -- from the West Indies and Africa -- will, over time, more strongly identify with the native-born African-American population or with the majority white population," Lichter said. "There also is a growing literature on ethnic antagonisms between native-born blacks and African-born immigrants. Hence our interest in intermarriage among diverse black populations -- intermarriage reveals important information on inter-group relations."
The importance of the study comes into sharper focus when viewed in a historical context. Miscegenation laws forbidding interracial marriage were not lifted nationwide until 1967, the study states. Even so, when it comes to marriage, the color line remains strong. Despite an increase in black-white marriages and cohabitation since 1967, African-Americans "remain much less likely than American Indians, Latinos and Asian-Americans to marry whites," Lichter said.
Skin color, then, remains a powerful and mitigating factor in America's marriage patterns.
July 26, 2006
Genes aren't the only code in DNA
This may prove to be a significant development. Read Nicholas Wade in the New York Times.
Researchers believe they have found a second code in DNA in addition to the genetic code.
The genetic code specifies all the proteins that a cell makes. The second code, superimposed on the first, sets the placement of the nucleosomes, miniature protein spools around which the DNA is looped. The spools both protect and control access to the DNA itself.
July 25, 2006
Moralistic punishment in front of an audience
Evolution and Human Behavior (Article in Press)
Audience effects on moralistic punishmentstar
Robert Kurzban et al.
Abstract
Punishment has been proposed as being central to two distinctively human phenomena: cooperation in groups and morality. Here we investigate moralistic punishment, a behavior designed to inflict costs on another individual in response to a perceived moral violation. There is currently no consensus on which evolutionary model best accounts for this phenomenon in humans. Models that turn on individuals' cultivating reputations as moralistic punishers clearly predict that psychological systems should be designed to increase punishment in response to information that one's decisions to punish will be known by others. We report two experiments in which we induce participants to commit moral violations and then present third parties with the opportunity to pay to punish wrongdoers. Varying conditions of anonymity, we find that the presence of an audience—even if only the experimenter—causes an increase in moralistic punishment.
Link
Audience effects on moralistic punishmentstar
Robert Kurzban et al.
Abstract
Punishment has been proposed as being central to two distinctively human phenomena: cooperation in groups and morality. Here we investigate moralistic punishment, a behavior designed to inflict costs on another individual in response to a perceived moral violation. There is currently no consensus on which evolutionary model best accounts for this phenomenon in humans. Models that turn on individuals' cultivating reputations as moralistic punishers clearly predict that psychological systems should be designed to increase punishment in response to information that one's decisions to punish will be known by others. We report two experiments in which we induce participants to commit moral violations and then present third parties with the opportunity to pay to punish wrongdoers. Varying conditions of anonymity, we find that the presence of an audience—even if only the experimenter—causes an increase in moralistic punishment.
Link
July 23, 2006
Hungry men like chubby women
Br J Psychol. 2006 Aug;97(Pt 3):353-63.
Does hunger influence judgments of female physical attractiveness?
Swami V, Tovee MJ.
To account for male preferences for female body weight following a consistent socio-economic pattern, Nelson and Morrison (2005) proposed a social-cognitive model based on the individual experience of resource scarcity. We replicated their studies showing that calorific dissatisfaction can influence preference for female body weight using a different dependent variable, namely photographic stimuli of women with known body weight and shape. Using this revised methodology, we found that operationalized intra-individual resource scarcity affects preferences for body weight: 30 hungry male participants preferred figures with a higher body weight and rated as more attractive heavier figures than 31 satiated male participants. Hungrier men were also less likely to be influenced by cues for body shape, supporting extant cross-cultural studies on female physical attractiveness. These findings corroborate those of Nelson and Morrison (2005) and are discussed in terms of how cultural contexts shape individual psychological experience as predicted by the theory of mutual constitution.
Link
Does hunger influence judgments of female physical attractiveness?
Swami V, Tovee MJ.
To account for male preferences for female body weight following a consistent socio-economic pattern, Nelson and Morrison (2005) proposed a social-cognitive model based on the individual experience of resource scarcity. We replicated their studies showing that calorific dissatisfaction can influence preference for female body weight using a different dependent variable, namely photographic stimuli of women with known body weight and shape. Using this revised methodology, we found that operationalized intra-individual resource scarcity affects preferences for body weight: 30 hungry male participants preferred figures with a higher body weight and rated as more attractive heavier figures than 31 satiated male participants. Hungrier men were also less likely to be influenced by cues for body shape, supporting extant cross-cultural studies on female physical attractiveness. These findings corroborate those of Nelson and Morrison (2005) and are discussed in terms of how cultural contexts shape individual psychological experience as predicted by the theory of mutual constitution.
Link
July 22, 2006
mtDNA and human movements in Oceania
Definitions of Remote Oceania and Near Oceania.
Mol Biol Evol. 2006 Jul 19; [Epub ahead of print]
Deciphering Past Human Population Movements in Oceania: Provably Optimal Trees of 127 mtDNA Genomes.
Pierson MJ, Martinez-Arias R, Holland BR, Gemmell NJ, Hurles ME, Penny D.
The settlement of the many island groups of Remote Oceania occurred relatively late in prehistory, beginning ca. 3000 years ago when people sailed eastwards into the Pacific from Near Oceania where evidence of human settlement dates from as early as 40 000 years ago. Archaeological and linguistic analyses have suggested the settlers of Remote Oceania had ancestry in Taiwan, as descendants of a proposed Neolithic expansion that began ca. 5500 years ago. Other researchers have suggested that the settlers were descendants of peoples from Island Southeast Asia, or the existing inhabitants of Near Oceania alone. To explore patterns of maternal descent in Oceania we have assembled and analyzed a dataset of 137 mtDNA genomes from Oceania, Australia, Island Southeast Asia and Taiwan that includes 19 sequences generated for this project. Using the MinMax Squeeze Approach (MMS) we report the consensus network of 165 most parsimonious trees for the Oceanic dataset, increasing by many orders of magnitude the numbers of trees for which a provable minimal solution has been found. The new mtDNA sequences highlight the limitations of partial sequencing for assigning sequences to haplogroups and dating recent divergence events. The provably optimal trees found for the entire mtDNA sequences using the MMS method provide a reliable and robust framework for the interpretation of evolutionary relationships and confirm the female settlers of Remote Oceania were descended from both the existing inhabitants of Near Oceania and more recent migrants into the region.
Link
Mol Biol Evol. 2006 Jul 19; [Epub ahead of print]
Deciphering Past Human Population Movements in Oceania: Provably Optimal Trees of 127 mtDNA Genomes.
Pierson MJ, Martinez-Arias R, Holland BR, Gemmell NJ, Hurles ME, Penny D.
The settlement of the many island groups of Remote Oceania occurred relatively late in prehistory, beginning ca. 3000 years ago when people sailed eastwards into the Pacific from Near Oceania where evidence of human settlement dates from as early as 40 000 years ago. Archaeological and linguistic analyses have suggested the settlers of Remote Oceania had ancestry in Taiwan, as descendants of a proposed Neolithic expansion that began ca. 5500 years ago. Other researchers have suggested that the settlers were descendants of peoples from Island Southeast Asia, or the existing inhabitants of Near Oceania alone. To explore patterns of maternal descent in Oceania we have assembled and analyzed a dataset of 137 mtDNA genomes from Oceania, Australia, Island Southeast Asia and Taiwan that includes 19 sequences generated for this project. Using the MinMax Squeeze Approach (MMS) we report the consensus network of 165 most parsimonious trees for the Oceanic dataset, increasing by many orders of magnitude the numbers of trees for which a provable minimal solution has been found. The new mtDNA sequences highlight the limitations of partial sequencing for assigning sequences to haplogroups and dating recent divergence events. The provably optimal trees found for the entire mtDNA sequences using the MMS method provide a reliable and robust framework for the interpretation of evolutionary relationships and confirm the female settlers of Remote Oceania were descended from both the existing inhabitants of Near Oceania and more recent migrants into the region.
Link
July 20, 2006
To my readers
I started this blog to keep some thoughts and references organized; being useful to others was only a secondary motivation. However, I have to admit that a picture like this does bring a smile to my face. So, thank you, readers, wherever you are!


July 18, 2006
How the Anglo-Saxons outbred the British
From an IOL story:
Proc. R. Soc. B doi:10.1098/rspb.2006.3627
Evidence for an apartheid-like social structure in early Anglo-Saxon England
Mark G. Thomas et al.
The role of migration in the Anglo-Saxon transition in England remains controversial. Archaeological and historical evidence is inconclusive, but current estimates of the contribution of migrants to the English population range from less than 10 000 to as many as 200 000. In contrast, recent studies based on Y-chromosome variation posit a considerably higher contribution to the modern English gene pool (50–100%). Historical evidence suggests that following the Anglo-Saxon transition, people of indigenous ethnicity were at an economic and legal disadvantage compared to those having Anglo-Saxon ethnicity. It is likely that such a disadvantage would lead to differential reproductive success. We examine the effect of differential reproductive success, coupled with limited intermarriage between distinct ethnic groups, on the spread of genetic variants. Computer simulations indicate that a social structure limiting intermarriage between indigenous Britons and an initially small Anglo-Saxon immigrant population provide a plausible explanation of the high degree of Continental male-line ancestry in England.
Link (pdf)
Britain - The Anglo-Saxons who conquered England in the fifth century set up a system of apartheid that enabled them to master and outbreed the native British majority, according to gene research published on Wednesday.UPDATE: Here is an audiovisual presentation based on a previous paper about Anglo-Saxon mass migration from the same team.
In less than 15 generations, more than half of the population in England had the genes of the invaders, investigators say.
"The native Britons were genetically and culturally absorbed by the Anglo-Saxons over a period of as little as a few hundred years," said Mark Thomas, a University College London biologist.
"An initially small invading Anglo-Saxon elite could have quickly established themselves by having more children who survived to adulthood, thanks to their military power and economic advantage.
"We believe that they also prevented the native British genes getting into the Anglo-Saxon population by restricting intermarriage in a system of apartheid that left the country culturally and genetically Germanised," he said.
"This is what we see today - a population of largely Germanic genetic origin, speaking a principally German language."
Proc. R. Soc. B doi:10.1098/rspb.2006.3627
Evidence for an apartheid-like social structure in early Anglo-Saxon England
Mark G. Thomas et al.
The role of migration in the Anglo-Saxon transition in England remains controversial. Archaeological and historical evidence is inconclusive, but current estimates of the contribution of migrants to the English population range from less than 10 000 to as many as 200 000. In contrast, recent studies based on Y-chromosome variation posit a considerably higher contribution to the modern English gene pool (50–100%). Historical evidence suggests that following the Anglo-Saxon transition, people of indigenous ethnicity were at an economic and legal disadvantage compared to those having Anglo-Saxon ethnicity. It is likely that such a disadvantage would lead to differential reproductive success. We examine the effect of differential reproductive success, coupled with limited intermarriage between distinct ethnic groups, on the spread of genetic variants. Computer simulations indicate that a social structure limiting intermarriage between indigenous Britons and an initially small Anglo-Saxon immigrant population provide a plausible explanation of the high degree of Continental male-line ancestry in England.
Link (pdf)
Origin of the Yakuts
Hum Genet. 2006 Jul 15; [Epub ahead of print]
Investigating the effects of prehistoric migrations in Siberia: genetic variation and the origins of Yakuts.
Pakendorf B, Novgorodov IN, Osakovskij VL, Danilova AP, Protod'jakonov AP, Stoneking M.
The Yakuts (also known as Sakha), Turkic-speaking cattle- and horse-breeders, inhabit a vast territory in Central and northeastern Siberia. On the basis of the archaeological, ethnographic and linguistic evidence, they are assumed to have migrated north from their original area of settlement in the vicinity of Lake Baykal in South Siberia under the pressure of the Mongol expansion during the thirteenth to fifteenth century AD: . During their initial migration and subsequent expansion, the ancestors of the Yakuts settled in the territory originally occupied by Tungusic- and Uralic-speaking reindeer-herders and hunters. In this paper we use mtDNA and Y-chromosomal analyses to elucidate whether the Yakut immigration and expansion was accompanied by admixture with the indigenous populations of their new area of settlement or whether the Yakuts displaced the original inhabitants without intermarriage. The mtDNA results show a very close affinity of the Yakuts with Central Asian and South Siberian groups, which confirms their southern origin. There is no conclusive evidence for admixture with indigenous populations, though a small amount cannot be excluded on the basis of the mtDNA data alone. The Y-chromosomal results confirm previous findings of a very strong bottleneck in the Yakuts, the age of which is in good accordance with the hypothesis that the Yakuts migrated north under Mongol pressure. Furthermore, the genetic results show that the Yakuts are a very homogenous population, notwithstanding their current spread over a very large territory. This confirms the historical accounts that they spread over their current area of settlement fairly recently.
Link
Investigating the effects of prehistoric migrations in Siberia: genetic variation and the origins of Yakuts.
Pakendorf B, Novgorodov IN, Osakovskij VL, Danilova AP, Protod'jakonov AP, Stoneking M.
The Yakuts (also known as Sakha), Turkic-speaking cattle- and horse-breeders, inhabit a vast territory in Central and northeastern Siberia. On the basis of the archaeological, ethnographic and linguistic evidence, they are assumed to have migrated north from their original area of settlement in the vicinity of Lake Baykal in South Siberia under the pressure of the Mongol expansion during the thirteenth to fifteenth century AD: . During their initial migration and subsequent expansion, the ancestors of the Yakuts settled in the territory originally occupied by Tungusic- and Uralic-speaking reindeer-herders and hunters. In this paper we use mtDNA and Y-chromosomal analyses to elucidate whether the Yakut immigration and expansion was accompanied by admixture with the indigenous populations of their new area of settlement or whether the Yakuts displaced the original inhabitants without intermarriage. The mtDNA results show a very close affinity of the Yakuts with Central Asian and South Siberian groups, which confirms their southern origin. There is no conclusive evidence for admixture with indigenous populations, though a small amount cannot be excluded on the basis of the mtDNA data alone. The Y-chromosomal results confirm previous findings of a very strong bottleneck in the Yakuts, the age of which is in good accordance with the hypothesis that the Yakuts migrated north under Mongol pressure. Furthermore, the genetic results show that the Yakuts are a very homogenous population, notwithstanding their current spread over a very large territory. This confirms the historical accounts that they spread over their current area of settlement fairly recently.
Link
July 13, 2006
New paper on haplogroup E-M78
I had written about this research about a year ago, and now the relevant paper has appeared in Human Mutation. The new phylogeny within haplogroup E-M78 is shown below:
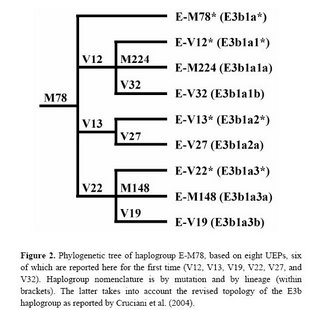
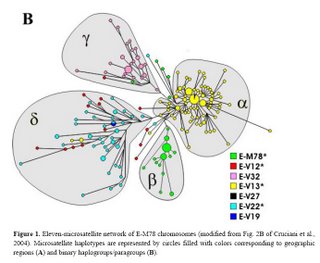
The newly discovered binary markers correspond roughly to the previously known microsatellite networks, although the correspondence is not perfect (see below):
From the paper:
Hum Mutat. 2006 Jul 11;27(8):831-832 [Epub ahead of print]
Molecular dissection of the Y chromosome haplogroup E-M78 (E3b1a): a posteriori evaluation of a microsatellite-network-based approach through six new biallelic markers.
Cruciani F, La Fratta R, Torroni A, Underhill PA, Scozzari R.
The human Y chromosome haplogroup E-M78 (E3b1a) occurs commonly and is distributed in northern and eastern Africa, western Asia, and all of Europe. Previously, only two rarely observed internal biallelic markers (UEPs) were known within the E-M78 clade. Here we report the identification of six novel UEPs that significantly refine the phylogeny of this haplogroup. Then, we evaluate the correspondence between the newly defined sub-haplogroups and the E-M78 haplotype clusters previously identified by an 11-microsatellite loci-based network encompassing 232 chromosomes (Cruciani et al., 2004). We observed considerable correspondence between the trees generated by the two types of markers, but also noted important discrepancies between microsatellite and UEP findings. Overall, this analysis reveals that the currently visible terminal branches of the Y tree still contain a large amount of information, in terms of undiscovered biallelic markers, and that caution is needed when using the microsatellite alleles as surrogates of unique event polymorphisms.
Link
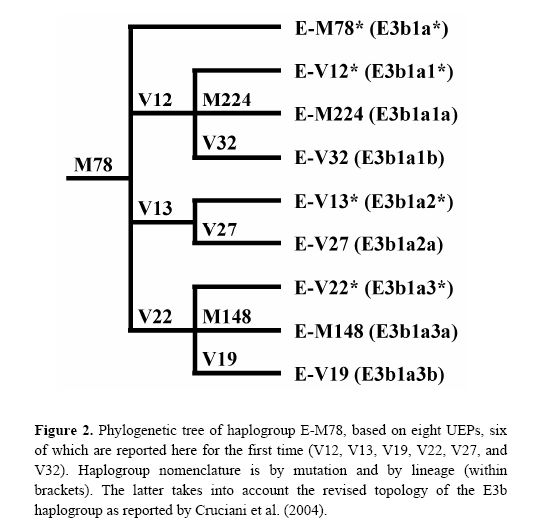
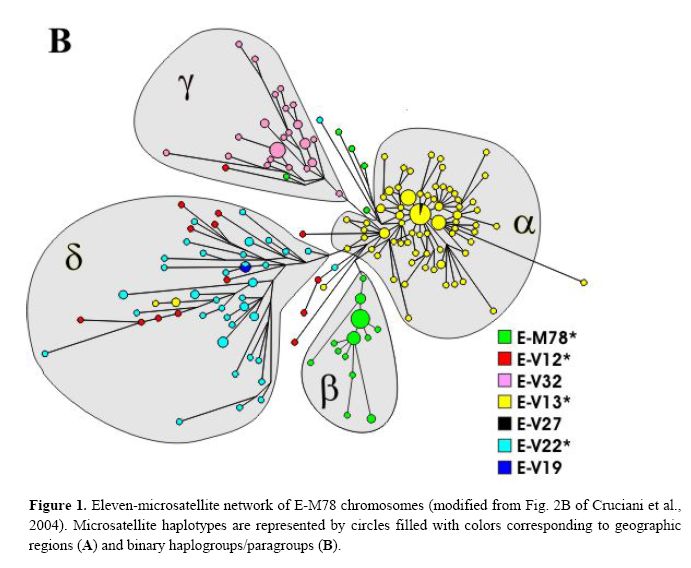
The newly discovered binary markers correspond roughly to the previously known microsatellite networks, although the correspondence is not perfect (see below):
From the paper:
Genetic differentiation among UEP defined groups of microsatellite-haplotypes is quite high (Rst = 0.59; P<10-3),> We also explored the correspondence between the two types of markers at a geographical level. There is a high and significant correlation observed between pairwise binary-haplogroup-based and microsatellite-haplotype-based distances among nine geographic regions (r = 0.91; P< 10-3). The two-dimensional plots from MDS show similar clusters of populations (Fig. 3), with slight differences possibly due to differential sensitivity of Φst and Rst to different mutational processes. North-western Africa, eastern Africa and the Balkans are well separated from each other and also from the central cluster in both diagrams. The differences are explained to a large extent by: the haplotypes associated with paragroup E-M78* (predominantly observed in north-western Africa), haplogroup E-V32 (found almost exclusively in eastern Africa) and haplogroup E-V13 (the only subset of E-M78 observed in the Balkans). E-V13 is also commonly found in the populations of the central cluster, where, however, other E-M78 sub-haplogroups are also present.
...
Taking into account the above data, the previously described European cluster α and the northern African cluster β are indeed confirmed as monophyletic groups of chromosomes, that, very likely, have their own defining binary marker yet to be discovered. Cluster α chromosomes constitute a major branch of the binary haplogroup V13, which, in turn, includes also a few, highly differentiated chromosomes - previously classified either in cluster δ or unclassified. All 29 chromosomes within cluster β belong to the paragroup E-M78*, which is relatively rare and almost exclusively restricted to a single geographic region (i.e. northern Africa), thus a common origin for at least a large part of these is likely.
Different scenarios characterize clusters δ and γ . The presence of three E-V13 chromosomes within cluster δ and the exclusion of some E-V12 and E-V22 chromosomes demonstrate that cluster δ cannot be regarded as a monophyletic unit. As for cluster γ, we have established close phylogenetic relationships of its members - now classified as E-V32 chromosomes - with those belonging to E-V12* within the E-V12 clade (Fig. 2). These relationships go undetected through the microsatellite network (red and pink chromosomes in Fig. 1B), most likely due to recurrent mutations at microsatellite loci. An alternative explanation would be that V12 has mutated twice independently in the E-M78 lineage. The latter possibility is less likely, given that V12 is within a terminal branch of the Y chromosome tree; moreover, it was never found by sequencing 18 Y*(xM78) chromosomes representing deep branches of the Y chromosome phylogeny (data not shown). Thus, the new markers we have detected now
offer the opportunity to explore in a better defined phylogenetic context the origin and distribution of the chromosomes belonging to haplogroup E-M78. Also, it is worth noting that twelve chromosomes that we were unable to assign to any cluster in the previous network analysis are now classified within four different haplogroups/paragroups (see Table 2 and Fig. 1B) as highly divergent microsatellite haplotypes. Thus, even though they represent only 5% of the total E-M78 chromosomes analyzed, their inclusion into the respective haplogroups/paragroups heavily affects inferences about time and place of origin of these haplogroups/paragroups.
Finally, although there is a strong correspondence between cluster γ − defined by the rare DYS19 11-repeat allele - and haplogroup E-V32, the presence, in cluster γ, of haplotypes belonging to the binary paragroups E M78* and E-V12* can only be explained by admitting either a paraphyletic or a polyphyletic origin for the chromosomes in the cluster.
Hum Mutat. 2006 Jul 11;27(8):831-832 [Epub ahead of print]
Molecular dissection of the Y chromosome haplogroup E-M78 (E3b1a): a posteriori evaluation of a microsatellite-network-based approach through six new biallelic markers.
Cruciani F, La Fratta R, Torroni A, Underhill PA, Scozzari R.
The human Y chromosome haplogroup E-M78 (E3b1a) occurs commonly and is distributed in northern and eastern Africa, western Asia, and all of Europe. Previously, only two rarely observed internal biallelic markers (UEPs) were known within the E-M78 clade. Here we report the identification of six novel UEPs that significantly refine the phylogeny of this haplogroup. Then, we evaluate the correspondence between the newly defined sub-haplogroups and the E-M78 haplotype clusters previously identified by an 11-microsatellite loci-based network encompassing 232 chromosomes (Cruciani et al., 2004). We observed considerable correspondence between the trees generated by the two types of markers, but also noted important discrepancies between microsatellite and UEP findings. Overall, this analysis reveals that the currently visible terminal branches of the Y tree still contain a large amount of information, in terms of undiscovered biallelic markers, and that caution is needed when using the microsatellite alleles as surrogates of unique event polymorphisms.
Link
July 11, 2006
Heritability of physical and psychological traits in 6,148 Sardinians
A new study in PLoS Genetics has estimated heritabilities of various traits over a large population of Sardinians. Please note that heritabilities calculated in one population should not be extended to other populations. If, e.g., there is a lot of environmental variation in nutrition in a population, then stature will be least affected by hereditary factors. Conversely, if all individuals are fed well, then the largest part of individual differences will be attributed to hereditary factors.
PLoS Genetics (Early Online Release)
Heritability of Cardiovascular and Personality Traits in 6,148 Sardinians
Giuseppe Pilia et al.
PROVISIONAL ABSTRACT
In large samples, phenotypic similarities between relatives yield information on the overall contribution of genes to trait variation. Large samples are especially important to compare heritability between subgroups such as young and old, or males and females. We recruited a cohort of 6,148 participants, aged 14-102, from 4 clustered towns in Sardinia. The cohort includes 34,469 relative pairs. To extract genetic information, we implemented software for variance components heritability analysis, designed to handle large pedigrees, analyze multiple traits simultaneously and model heterogeneity. Here, we report heritability analyses for 98 quantitative traits, focusing on facets of personality and cardiovascular function. We also summarize results of bivariate analyses for all pairs of traits and of heterogeneity analyses for each trait. We found a significant genetic component for every trait. On average genetic effects explained 40% of the variance for 38 blood tests, 51% for 5 anthropometric measures, 25% for 20 measures of cardiovascular function, and 19% for 35 personality traits. Four traits showed significant evidence for an X-linked component. Bivariate analyses suggested overlapping genetic determinants for many traits, including multiple personality facets and several traits related to the metabolic syndrome; but we found no evidence for shared genetic determinant that might underlie the reported association of some personality traits and cardiovascular risk factors. Models allowing for heterogeneity suggested that, in this cohort, the genetic variance was typically larger in females and in younger individuals, but interesting exceptions were observed. For example, narrow heritability of blood pressure was ~26% in individuals >42 years old, but only ~8% in younger individuals. Despite the heterogeneity in effect sizes, the same loci appear to contribute to variance in young and old and in males and females. In summary, we find significant evidence for heritability of many medically important traits, including cardiovascular function and personality. Evidence for heterogeneity by age and sex suggest that models allowing for these differences will be important in mapping quantitative traits.
Link
PLoS Genetics (Early Online Release)
Heritability of Cardiovascular and Personality Traits in 6,148 Sardinians
Giuseppe Pilia et al.
PROVISIONAL ABSTRACT
In large samples, phenotypic similarities between relatives yield information on the overall contribution of genes to trait variation. Large samples are especially important to compare heritability between subgroups such as young and old, or males and females. We recruited a cohort of 6,148 participants, aged 14-102, from 4 clustered towns in Sardinia. The cohort includes 34,469 relative pairs. To extract genetic information, we implemented software for variance components heritability analysis, designed to handle large pedigrees, analyze multiple traits simultaneously and model heterogeneity. Here, we report heritability analyses for 98 quantitative traits, focusing on facets of personality and cardiovascular function. We also summarize results of bivariate analyses for all pairs of traits and of heterogeneity analyses for each trait. We found a significant genetic component for every trait. On average genetic effects explained 40% of the variance for 38 blood tests, 51% for 5 anthropometric measures, 25% for 20 measures of cardiovascular function, and 19% for 35 personality traits. Four traits showed significant evidence for an X-linked component. Bivariate analyses suggested overlapping genetic determinants for many traits, including multiple personality facets and several traits related to the metabolic syndrome; but we found no evidence for shared genetic determinant that might underlie the reported association of some personality traits and cardiovascular risk factors. Models allowing for heterogeneity suggested that, in this cohort, the genetic variance was typically larger in females and in younger individuals, but interesting exceptions were observed. For example, narrow heritability of blood pressure was ~26% in individuals >42 years old, but only ~8% in younger individuals. Despite the heterogeneity in effect sizes, the same loci appear to contribute to variance in young and old and in males and females. In summary, we find significant evidence for heritability of many medically important traits, including cardiovascular function and personality. Evidence for heterogeneity by age and sex suggest that models allowing for these differences will be important in mapping quantitative traits.
Link
Paternity Confidence
I had written about a study quantifying the number of fathers who aren't really fathers of their children, even though they think they are. The study has now appeared in the latest issue of Current Anthropology
CURRENT ANTHROPOLOGY Volume 47, Number 3, June 2006
How Well Does Paternity Confidence Match Actual Paternity?
Evidence from Worldwide Nonpaternity Rates
Kermyt G. Anderson
Evolutionary theory predicts that males will provide less parental investment for putative offspring who are unlikely to be their actual offspring. Cross-culturally, paternity confidence (a man's assessment of the likelihood that he is the father of a putative child) is positively associated with men's involvement with children and with investment or inheritance from paternal kin. A survey of 67 studies reporting nonpaternity suggests that for men with high paternity confidence rates of nonpaternity are(excluding studies of unknown methodology) typically 1.9%, substantially less than the typical rates of 10% or higher cited by many researchers. Further cross-cultural investigation of the relationship between paternity and paternity confidence is warranted.
Link
CURRENT ANTHROPOLOGY Volume 47, Number 3, June 2006
How Well Does Paternity Confidence Match Actual Paternity?
Evidence from Worldwide Nonpaternity Rates
Kermyt G. Anderson
Evolutionary theory predicts that males will provide less parental investment for putative offspring who are unlikely to be their actual offspring. Cross-culturally, paternity confidence (a man's assessment of the likelihood that he is the father of a putative child) is positively associated with men's involvement with children and with investment or inheritance from paternal kin. A survey of 67 studies reporting nonpaternity suggests that for men with high paternity confidence rates of nonpaternity are(excluding studies of unknown methodology) typically 1.9%, substantially less than the typical rates of 10% or higher cited by many researchers. Further cross-cultural investigation of the relationship between paternity and paternity confidence is warranted.
Link
July 09, 2006
Les Bleus vs. Squadra Azzurra
World Cup 2006 will be decided soon...

See also, The geography of European phenotypical variation and Composites of Greek and European Female Athletes for more facial composites.

See also, The geography of European phenotypical variation and Composites of Greek and European Female Athletes for more facial composites.
July 07, 2006
The Homo floresiensis debate rages on
Well, the saga of the hobbit continues in a new article on the Journal of Human Evolution. Some previous entries on the topic. The new paper comes in favor of the idea that the Flores hominid represents a new species.
Journal of Human Evolution
Homo floresiensis: Microcephalic, pygmoid, Australopithecus, or Homo? (In Press, Accepted Manuscript, Available online 5 July 2006)
Debbie Argue, Denise Donlon, Colin Groves and Richard Wright
Abstract
The remarkable partial adult skeleton (LB1) excavated from Liang Bua cave on the island of Flores, Indonesia, has been attributed to a new species, Homo floresiensis, based upon a unique mosaic of primitive and derived features compared to any other hominin. The announcement precipitated widespread interest, and attention quickly focused on its possible affinities. LB1 is a small-bodied hominin with an endocranial volume of 380–410 cm3, a stature of 1 m, and an approximate geological age of 18,000 years. The describers (Brown et al., 2004) originally proposed that H. floresiensis was the end product of a long period of isolation of H. erectus or early Homo on a small island, a process known as insular dwarfism. More recently, Morwood, Brown, and colleagues (2005) reviewed this assessment in light of new material from the site and concluded that H. floresiensis is not likely to be descended from H. erectus, with the genealogy of the species remaining uncertain. Other interpretations, namely that LB1 is a pygmy or afflicted with microcephaly, have also been put forward.
We explore the affinities of LB1 using cranial and postcranial metric and nonmetric analyses. LB1 is compared to early Homo, two microcephalic humans, a ‘pygmoid’ excavated from another cave on Flores, H. sapiens (including African pygmies and Andaman Islanders), Australopithecus, and Paranthropus. Based on these comparisons, we conclude that it is unlikely that LB1 is a microcephalic human, and it cannot be attributed to any known species. Its attribution to a new species, Homo floresiensis, is supported.
Journal of Human Evolution
Homo floresiensis: Microcephalic, pygmoid, Australopithecus, or Homo? (In Press, Accepted Manuscript, Available online 5 July 2006)
Debbie Argue, Denise Donlon, Colin Groves and Richard Wright
Abstract
The remarkable partial adult skeleton (LB1) excavated from Liang Bua cave on the island of Flores, Indonesia, has been attributed to a new species, Homo floresiensis, based upon a unique mosaic of primitive and derived features compared to any other hominin. The announcement precipitated widespread interest, and attention quickly focused on its possible affinities. LB1 is a small-bodied hominin with an endocranial volume of 380–410 cm3, a stature of 1 m, and an approximate geological age of 18,000 years. The describers (Brown et al., 2004) originally proposed that H. floresiensis was the end product of a long period of isolation of H. erectus or early Homo on a small island, a process known as insular dwarfism. More recently, Morwood, Brown, and colleagues (2005) reviewed this assessment in light of new material from the site and concluded that H. floresiensis is not likely to be descended from H. erectus, with the genealogy of the species remaining uncertain. Other interpretations, namely that LB1 is a pygmy or afflicted with microcephaly, have also been put forward.
We explore the affinities of LB1 using cranial and postcranial metric and nonmetric analyses. LB1 is compared to early Homo, two microcephalic humans, a ‘pygmoid’ excavated from another cave on Flores, H. sapiens (including African pygmies and Andaman Islanders), Australopithecus, and Paranthropus. Based on these comparisons, we conclude that it is unlikely that LB1 is a microcephalic human, and it cannot be attributed to any known species. Its attribution to a new species, Homo floresiensis, is supported.
July 05, 2006
DNA contamination of ancient teeth
Genetic studies of ancient human populations usually rely on amplification of DNA preserved in dental samples. Two main problems plague such studies. First, DNA suffers damage after death and until it is studied by scientists. Therefore, it is not clear which parts of a DNA sequence are due to the genetic makeup of the ancient individual, and which ones have been modified after his/her death.
The second problem is due to the fact that skeletal remains are usually handled by many people, e.g., excavators, archaeologists, anthropologists, after they are taken off the ground. This may introduce the handlers' DNA on them. Then, when DNA is amplified, it is not clear whether the observed sequence is from the ancient individual or the more recent handlers. During recent excavations, people take care not to contaminate samples. Moreover, they tend to record meticulously the group of individuals handling the remains, since this is useful to check whether a DNA sequence may belong to one of them. Unfortunately, many useful ancient skeletons were excavated before such precautions were in place.
The identification of these two problems does not, in itself, quantify their severity. How much faith should we have in DNA sequences amplified from ancient teeth? This new paper is a welcome contribution to tackling this important problem.
Molecular Biology and Evolution (Advance Access)
Tracking Down Human Contamination in Ancient Human Teeth
María Lourdes Sampietro et al.
Abstract
DNA contamination arising from the manipulation of ancient calcified tissue samples is a poorly understood, yet fundamental problem that affects the reliability of ancient DNA studies. We have typed the mitochondrial DNA hypervariable region I (HVR1) of the only six people involved in the excavation, washing and subsequent anthropological and genetic study of 23 Neolithic remains excavated from Granollers (Barcelona, Spain), and examined for their presence among the 572 clones generated during the aDNA analyses of teeth from these samples. 17.13% of the cloned sequences could be unambiguously identified as contaminants, with those derived from the people involved in the retrieval and washing of the remains present in higher frequencies than those of the anthropologist and genetic researchers. This finding confirms for the first time previous hypotheses that teeth samples are most susceptible to contamination at their initial excavation. More worrying, the cloned contaminant sequences exhibit substitutions that can be attributed to DNA damage after the contamination event, and we demonstrate that the level of such damage increases with time; contaminants that are >10 years old have approximately five times more damage than those that are recent. Furthermore, we demonstrate that in this dataset, the damage rate of the old contaminant sequences is indistinguishable from that of the endogenous DNA sequences. As such, the commonly used argument that miscoding lesions observed among cloned aDNA sequences can be used to support data authenticity, is misleading in scenarios where the presence of old contaminant sequences is possible. We argue therefore that the typing of those involved in the manipulation of the ancient human specimens is critical in order to ensure that generated results are accurate.
Link
The second problem is due to the fact that skeletal remains are usually handled by many people, e.g., excavators, archaeologists, anthropologists, after they are taken off the ground. This may introduce the handlers' DNA on them. Then, when DNA is amplified, it is not clear whether the observed sequence is from the ancient individual or the more recent handlers. During recent excavations, people take care not to contaminate samples. Moreover, they tend to record meticulously the group of individuals handling the remains, since this is useful to check whether a DNA sequence may belong to one of them. Unfortunately, many useful ancient skeletons were excavated before such precautions were in place.
The identification of these two problems does not, in itself, quantify their severity. How much faith should we have in DNA sequences amplified from ancient teeth? This new paper is a welcome contribution to tackling this important problem.
Molecular Biology and Evolution (Advance Access)
Tracking Down Human Contamination in Ancient Human Teeth
María Lourdes Sampietro et al.
Abstract
DNA contamination arising from the manipulation of ancient calcified tissue samples is a poorly understood, yet fundamental problem that affects the reliability of ancient DNA studies. We have typed the mitochondrial DNA hypervariable region I (HVR1) of the only six people involved in the excavation, washing and subsequent anthropological and genetic study of 23 Neolithic remains excavated from Granollers (Barcelona, Spain), and examined for their presence among the 572 clones generated during the aDNA analyses of teeth from these samples. 17.13% of the cloned sequences could be unambiguously identified as contaminants, with those derived from the people involved in the retrieval and washing of the remains present in higher frequencies than those of the anthropologist and genetic researchers. This finding confirms for the first time previous hypotheses that teeth samples are most susceptible to contamination at their initial excavation. More worrying, the cloned contaminant sequences exhibit substitutions that can be attributed to DNA damage after the contamination event, and we demonstrate that the level of such damage increases with time; contaminants that are >10 years old have approximately five times more damage than those that are recent. Furthermore, we demonstrate that in this dataset, the damage rate of the old contaminant sequences is indistinguishable from that of the endogenous DNA sequences. As such, the commonly used argument that miscoding lesions observed among cloned aDNA sequences can be used to support data authenticity, is misleading in scenarios where the presence of old contaminant sequences is possible. We argue therefore that the typing of those involved in the manipulation of the ancient human specimens is critical in order to ensure that generated results are accurate.
Link